SecStrAnnotator:Analysis: Difference between revisions
No edit summary |
|||
| Line 4: | Line 4: | ||
- <code>secondary_structure_anatomy.R</code> – contains commands for reading the annotation results, generating plots, and performing some statistical test to compare eukaryotic and bacterial structures (or any two sets of structures) | - <code>secondary_structure_anatomy.R</code> – contains commands for reading the annotation results, generating plots, and performing some statistical test to compare eukaryotic and bacterial structures (or any two sets of structures) | ||
==Procedure== | |||
===Data preparation=== | |||
<code>scripts/secstrapi_data_preparation/SecStrAPI_master.sh</code> Contains a pipeline for annotating the whole protein family, including: | |||
- downloading the list of family members defined by CATH and Pfam, | |||
- downloading their structures, | |||
- selecting a non-redundant set, | |||
- annotation, | |||
- multiple sequence alignment for individual SSEs, | |||
- formatting into SecStrAPI format, | |||
- formatting into TSV format for further analyses. | |||
Example usage: | |||
bash scripts/secstrapi_data_preparation/SecStrAPI_master.sh | |||
Before running, modify the SETTINGS section of the SecStrAPI_master.sh to set your family of interest, data directory, options, paths to your template annotation (TEMPLATE_ANNOTATION_FILE, TEMPLATE_STRUCTURE_FILE). Unwanted steps of the pipeline can be commented out in the MAIN PIPELINE section. | |||
===Data analysis=== | |||
| Line 11: | Line 36: | ||
For the Cytochrome P450 family, structures of 1775 protein domains are available, located in 953 PDB entries. The analysis was performed on a non-redundant subset containing 175 protein domains. | For the Cytochrome P450 family, structures of 1775 protein domains are available, located in 953 PDB entries. The analysis was performed on a non-redundant subset containing 175 protein domains. | ||
===Occurrence of SSEs=== | ===Occurrence of SSEs=== | ||
Revision as of 10:54, 9 April 2020
SecStrAnnotator Suite provides scripts (Python, R, and bash) for batch annotation of the whole family and analysis of the annotation results. The main scripts are
- SecStrAPI_master.sh – this bash script used to prepare data for SecStrAPI also formats the annotations into tab-separated (TSV) files readable by R
- secondary_structure_anatomy.R – contains commands for reading the annotation results, generating plots, and performing some statistical test to compare eukaryotic and bacterial structures (or any two sets of structures)
Procedure
Data preparation
scripts/secstrapi_data_preparation/SecStrAPI_master.sh Contains a pipeline for annotating the whole protein family, including:
- downloading the list of family members defined by CATH and Pfam,
- downloading their structures,
- selecting a non-redundant set,
- annotation,
- multiple sequence alignment for individual SSEs,
- formatting into SecStrAPI format,
- formatting into TSV format for further analyses.
Example usage:
bash scripts/secstrapi_data_preparation/SecStrAPI_master.sh
Before running, modify the SETTINGS section of the SecStrAPI_master.sh to set your family of interest, data directory, options, paths to your template annotation (TEMPLATE_ANNOTATION_FILE, TEMPLATE_STRUCTURE_FILE). Unwanted steps of the pipeline can be commented out in the MAIN PIPELINE section.
Data analysis
Example case study: Cytochromes P450
Data
For the Cytochrome P450 family, structures of 1775 protein domains are available, located in 953 PDB entries. The analysis was performed on a non-redundant subset containing 175 protein domains.
Occurrence of SSEs
The occurrence describes in what percentage of the structures a particular SSE is present.
-
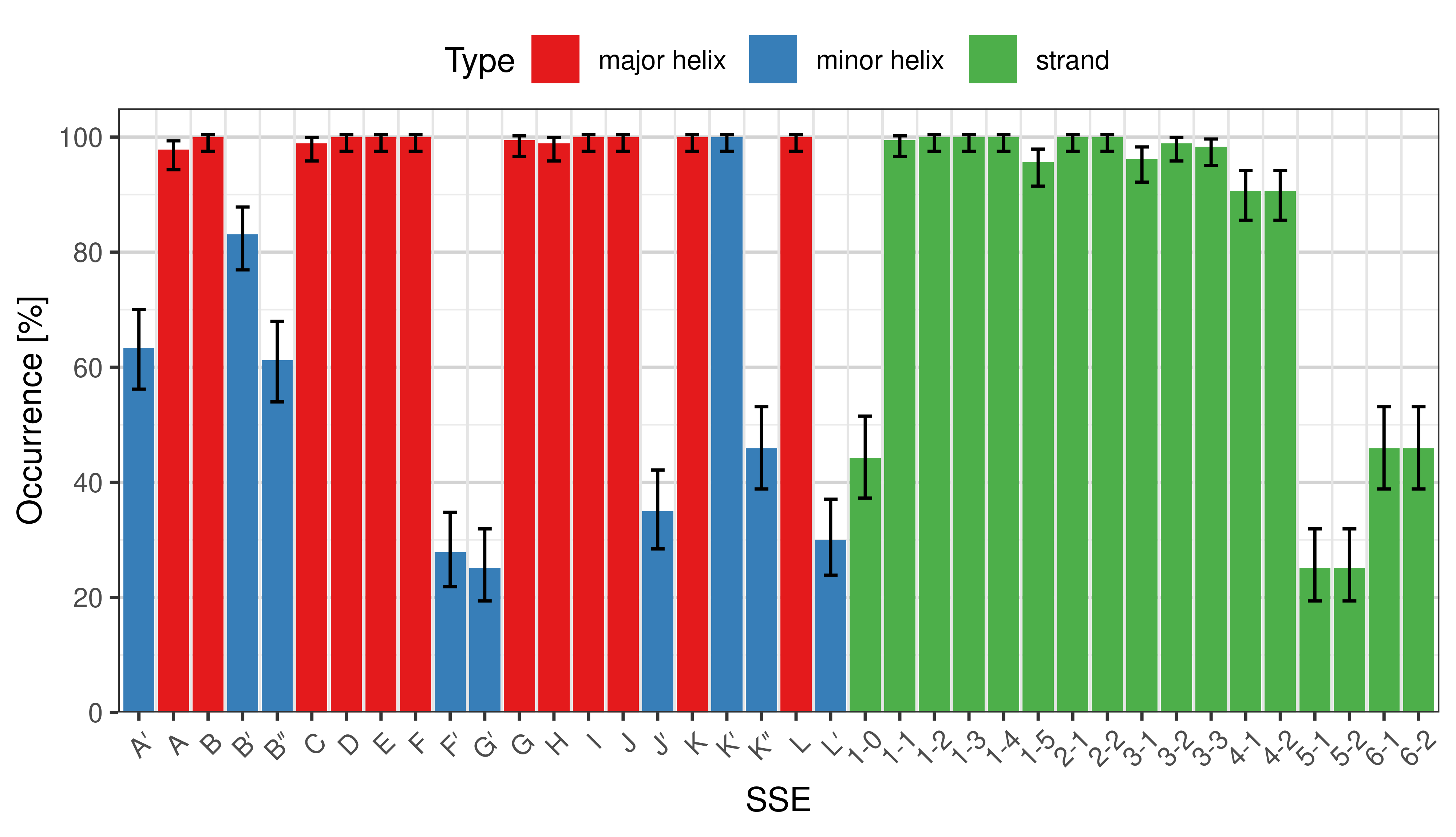
Occurrence of particular SSEs in the whole set. -
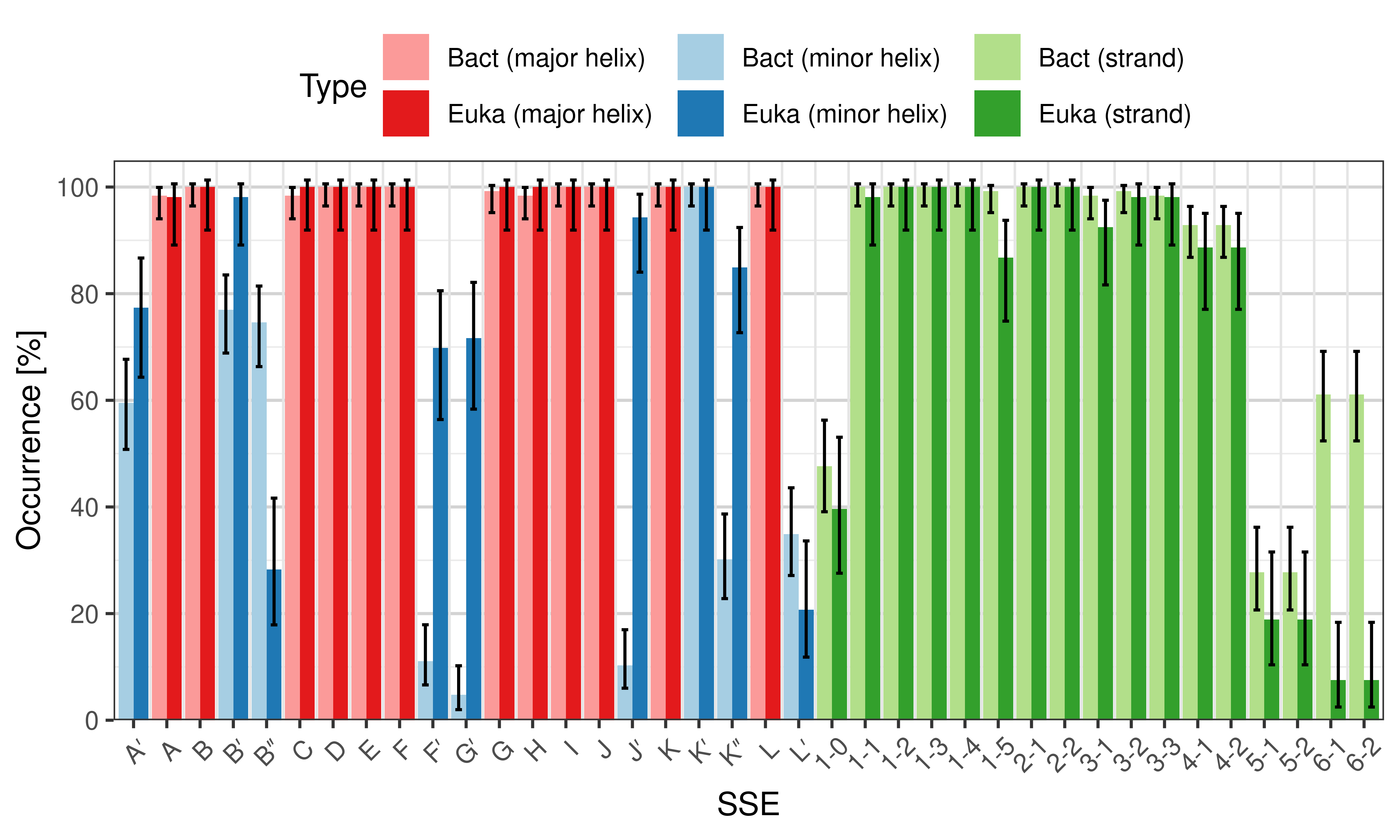
Occurrence of particular SSEs – comparison of bacterial and eukaryotic structures.


Length of SSEs
The length of an SSE is measured as the number of residues. The following violin plots show the distribution of length for each SSE.
-
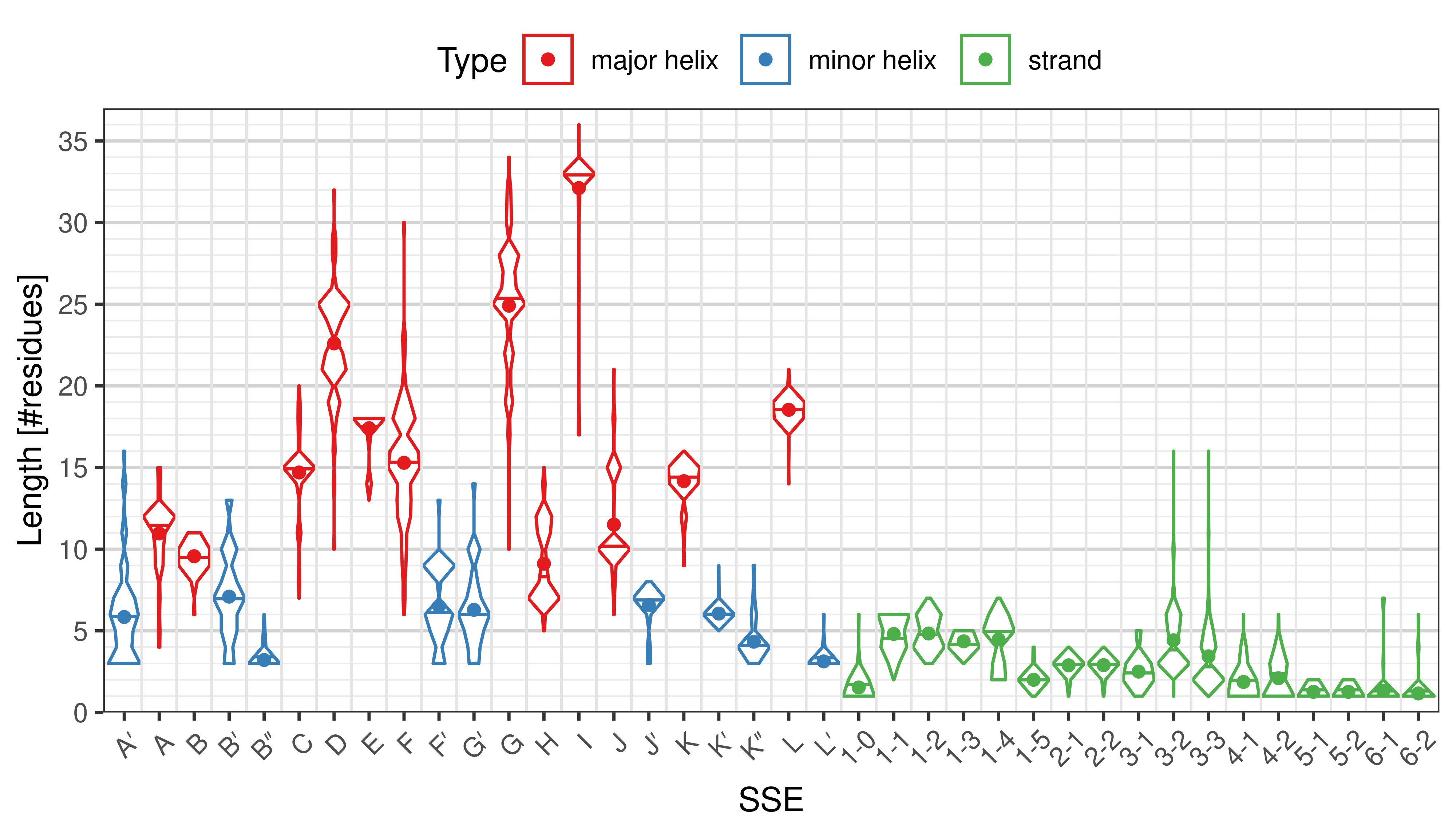
Length distribution of particular SSEs in the whole set. -
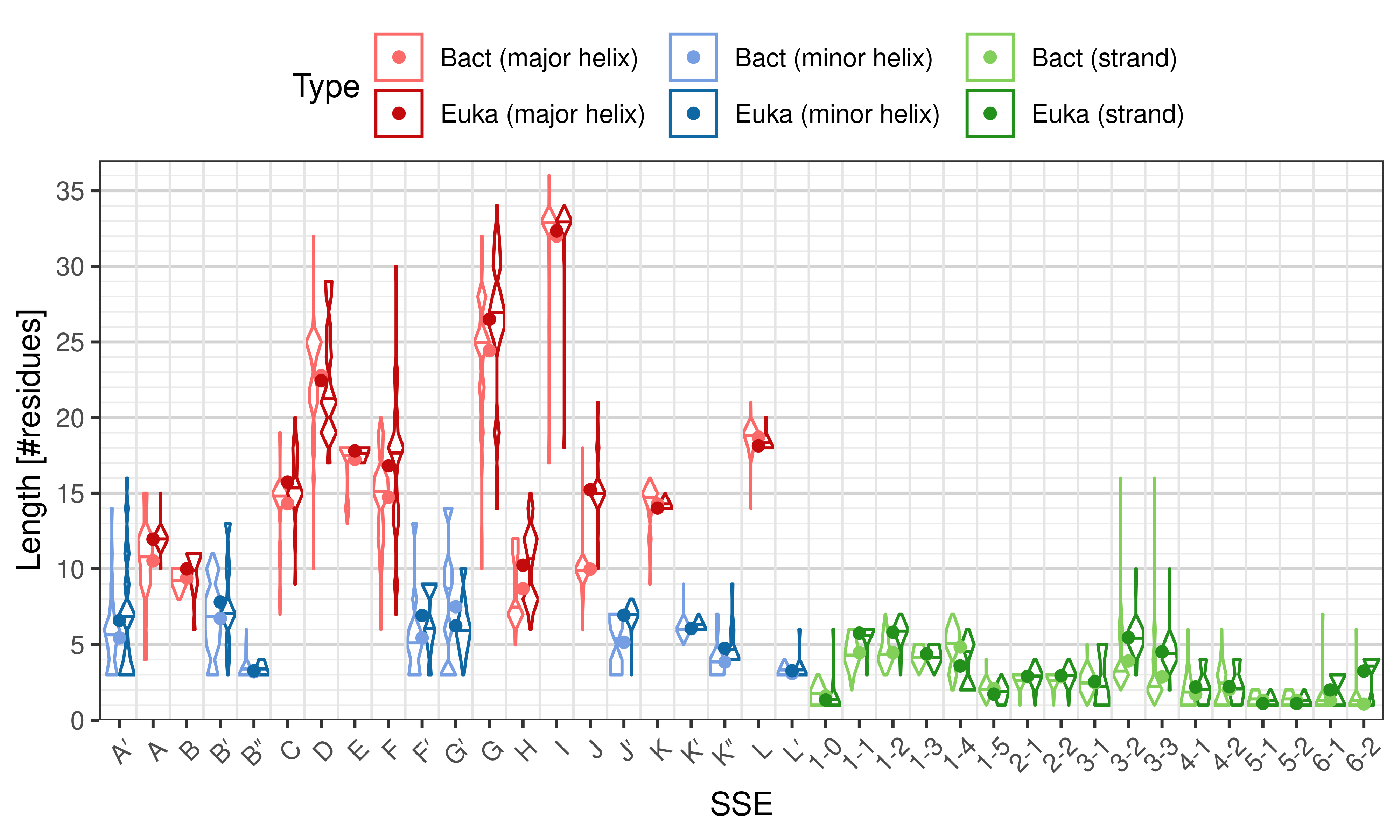
Length distribution of particular SSEs – comparison of bacterial and eukaryotic structures.


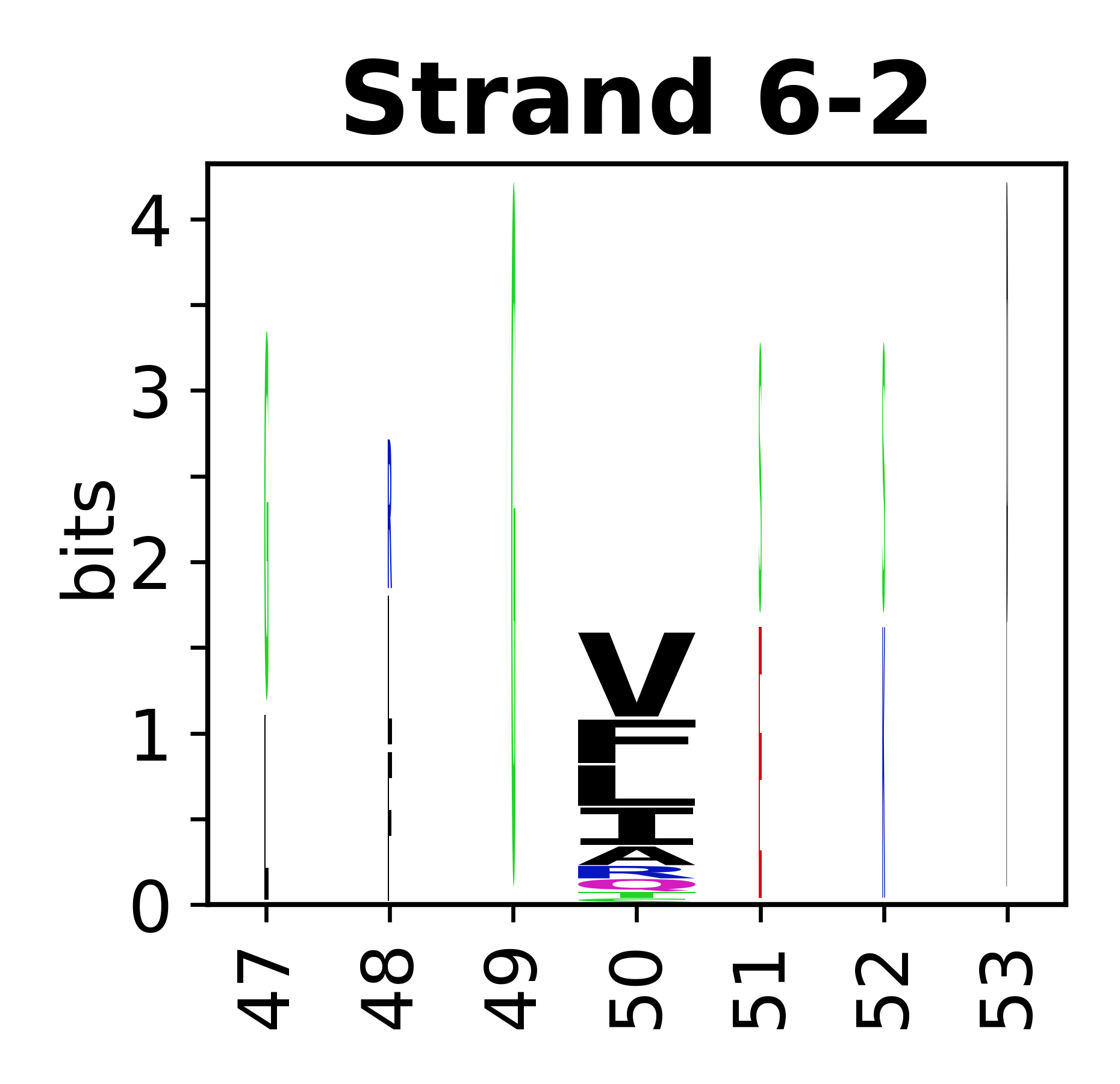
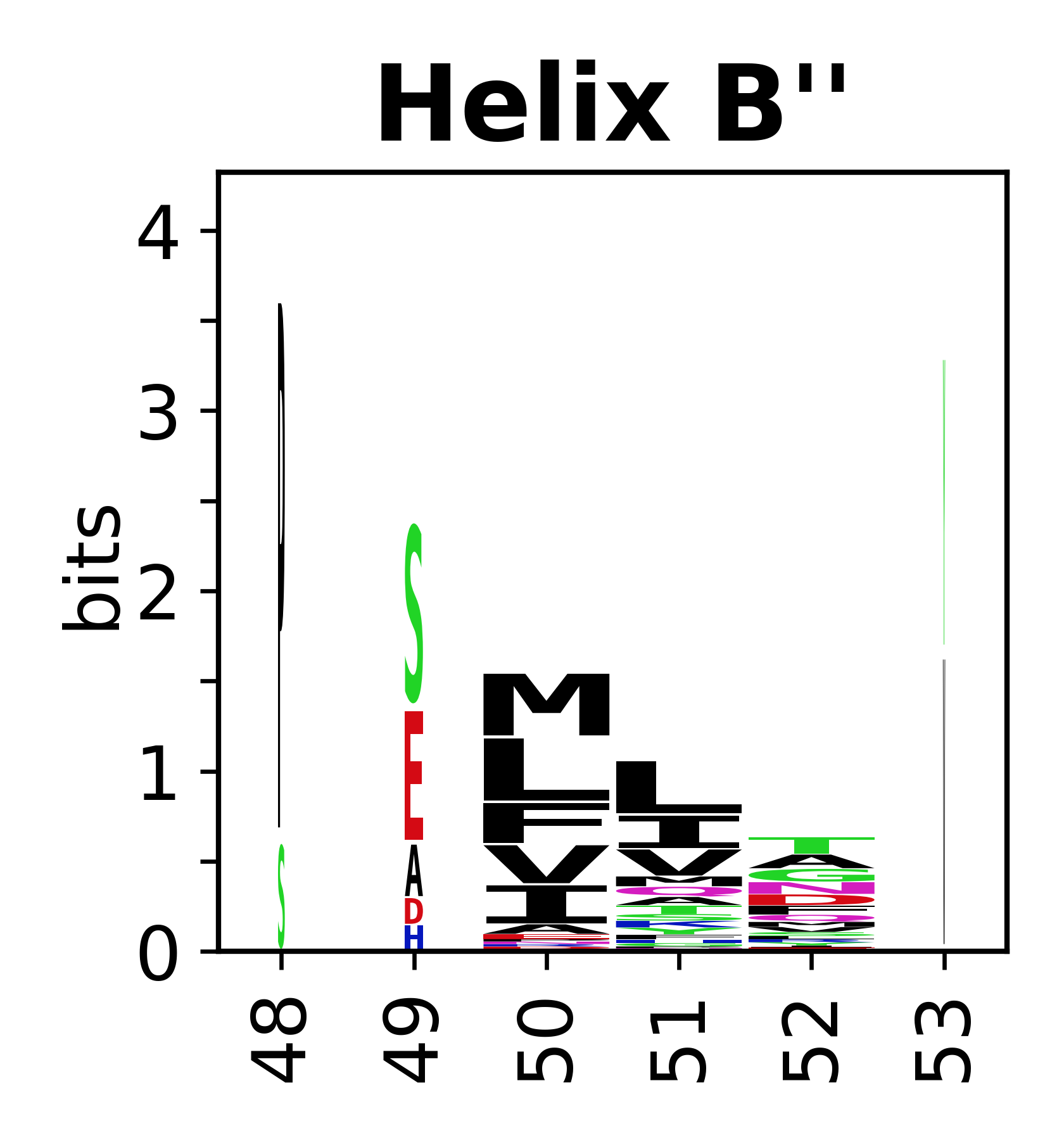
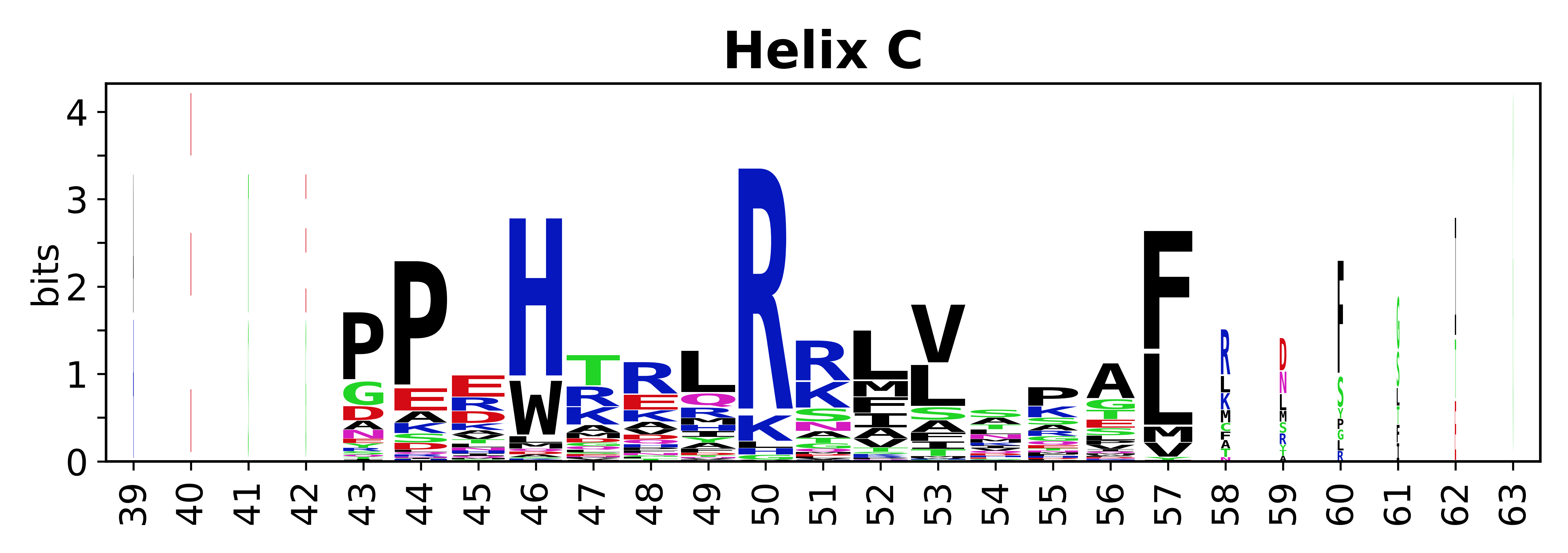
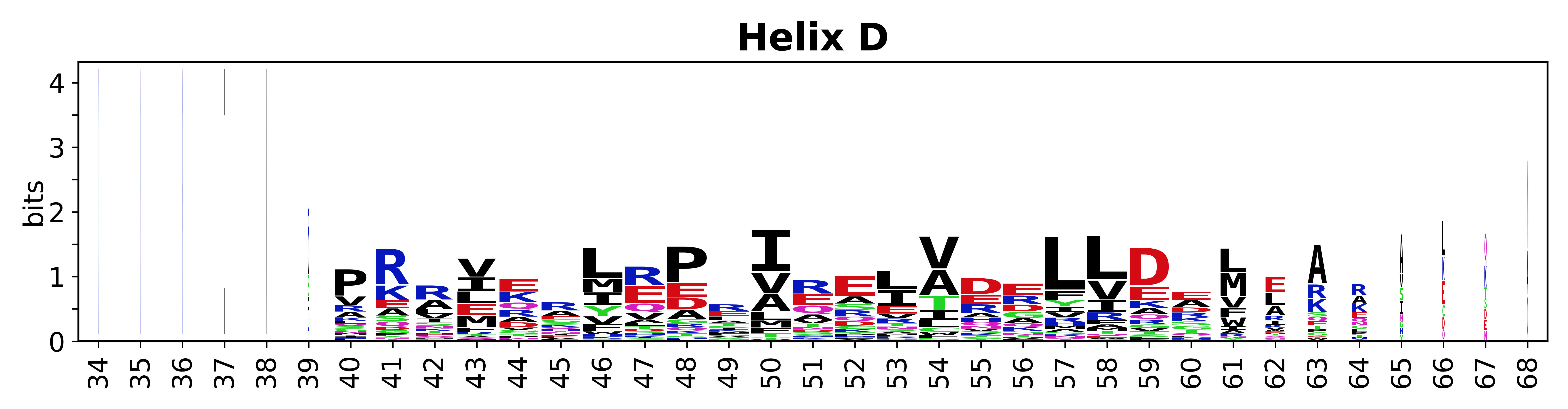
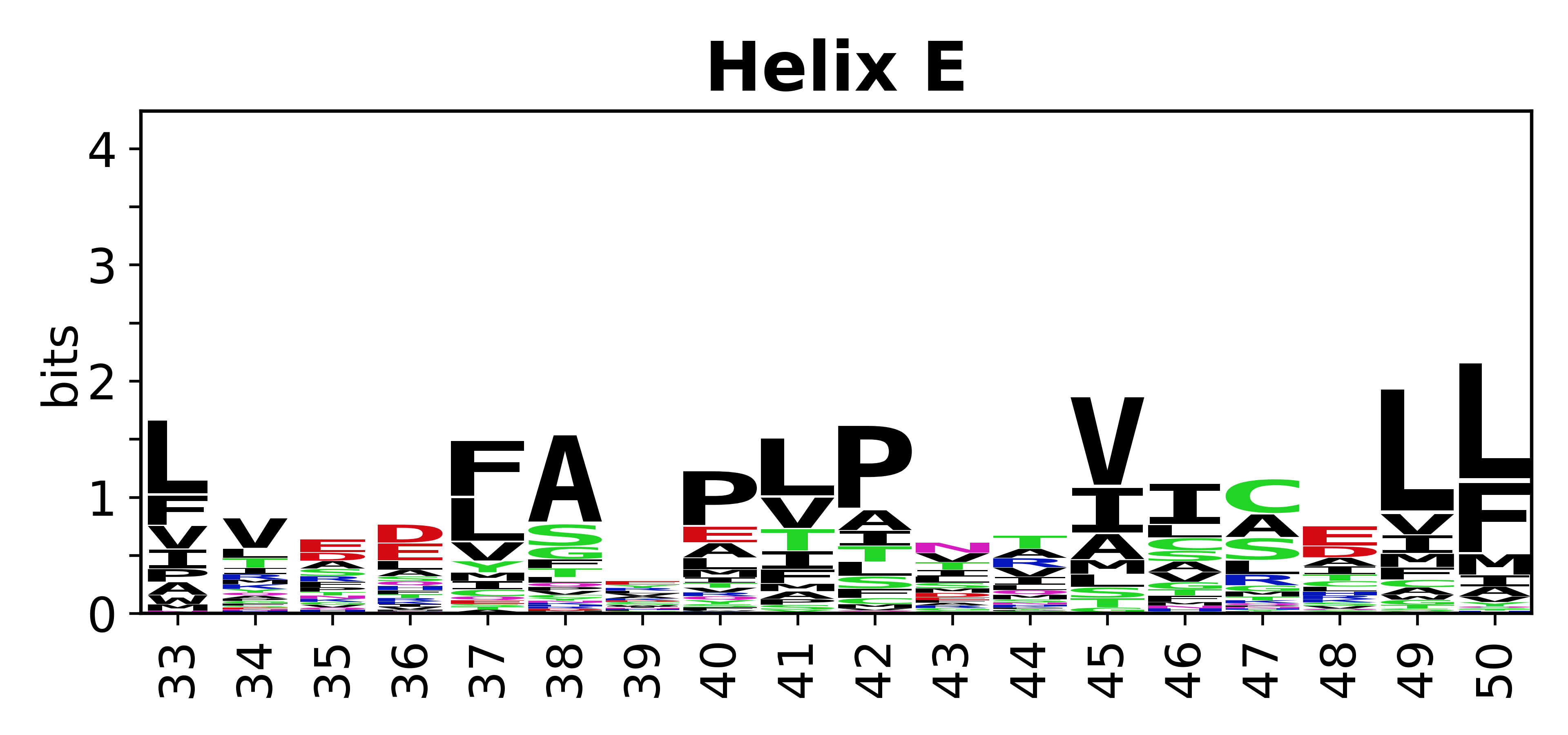
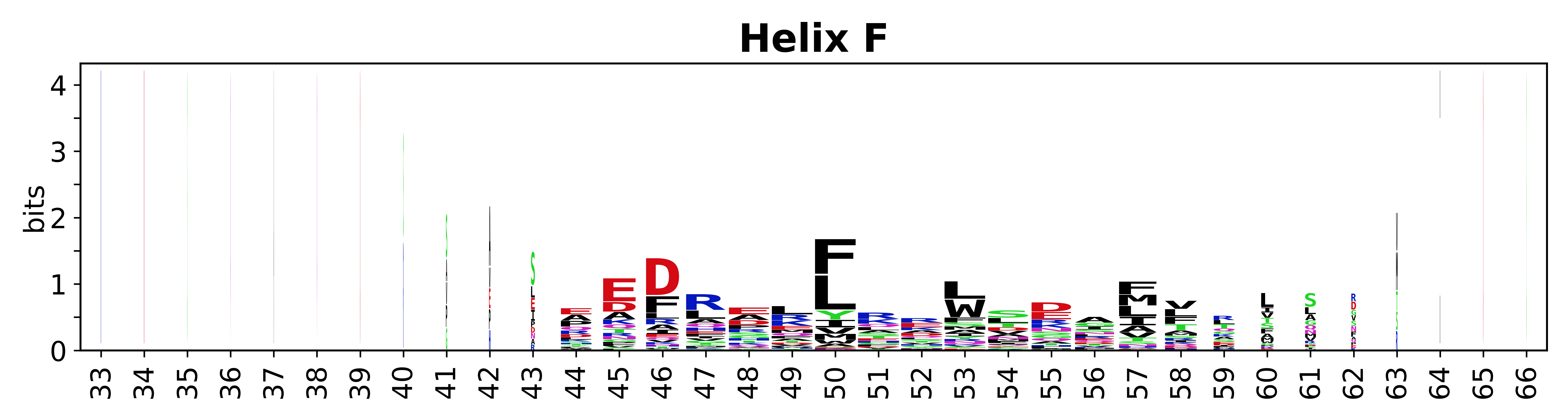
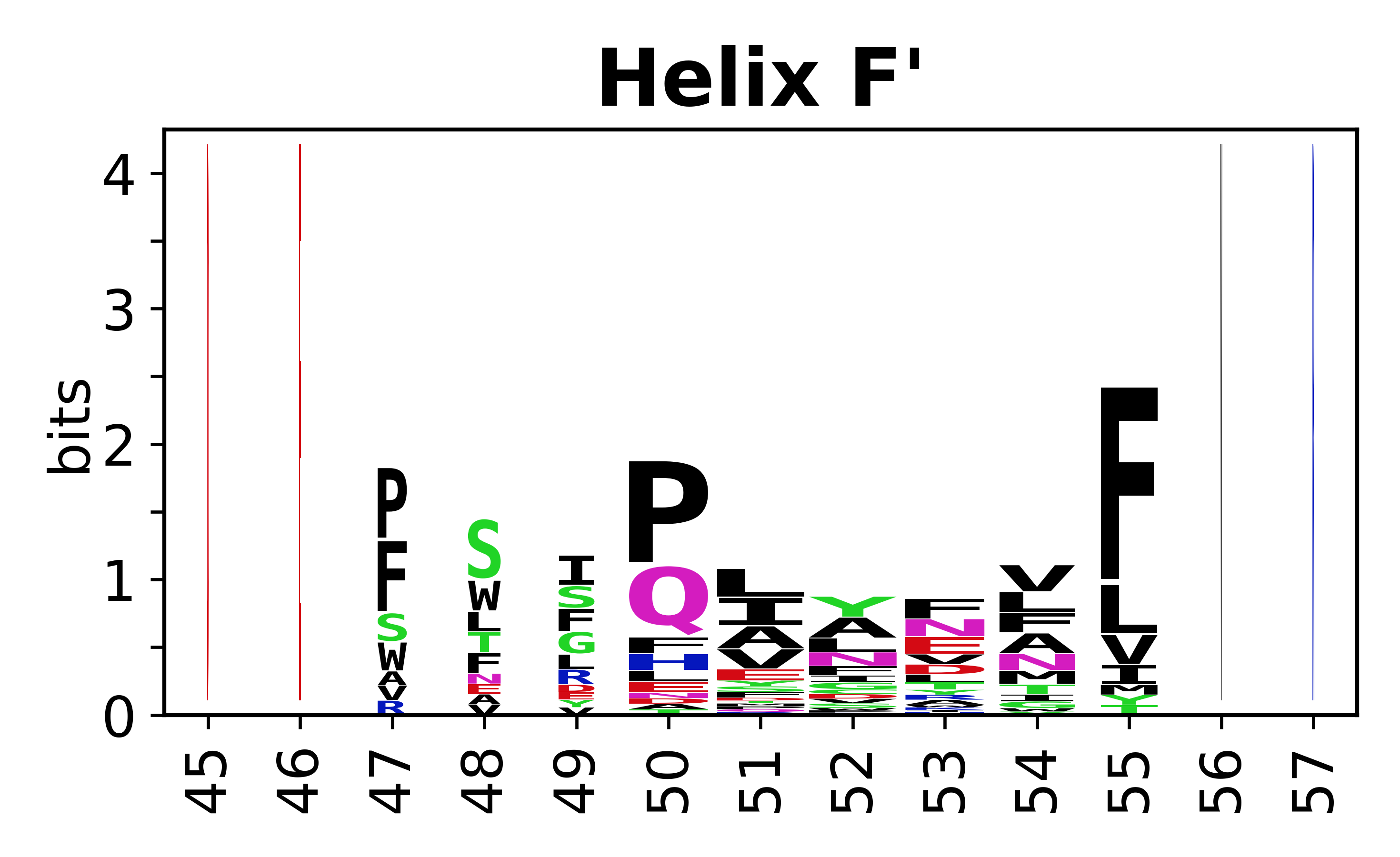
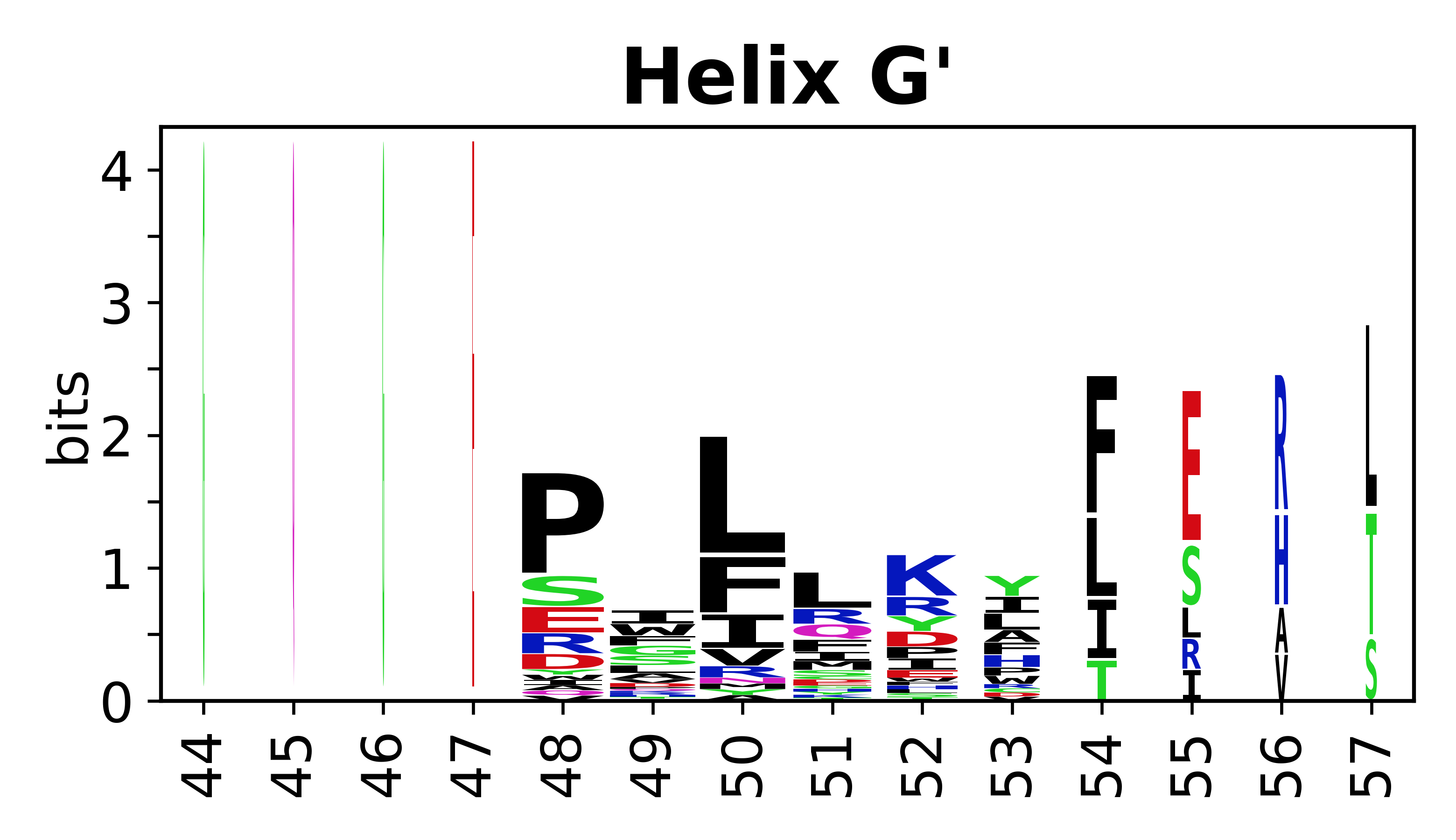
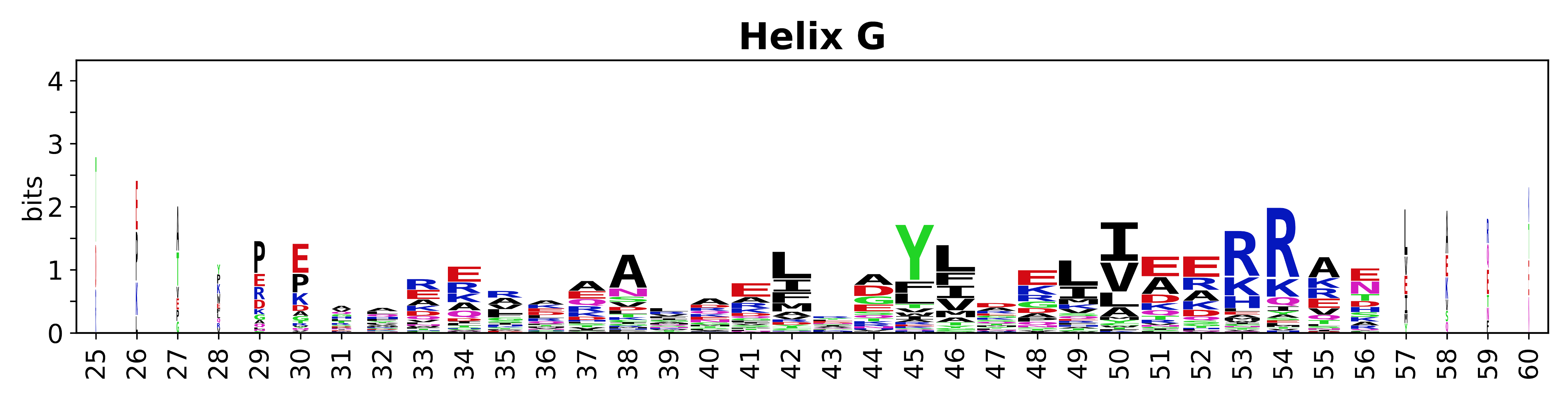
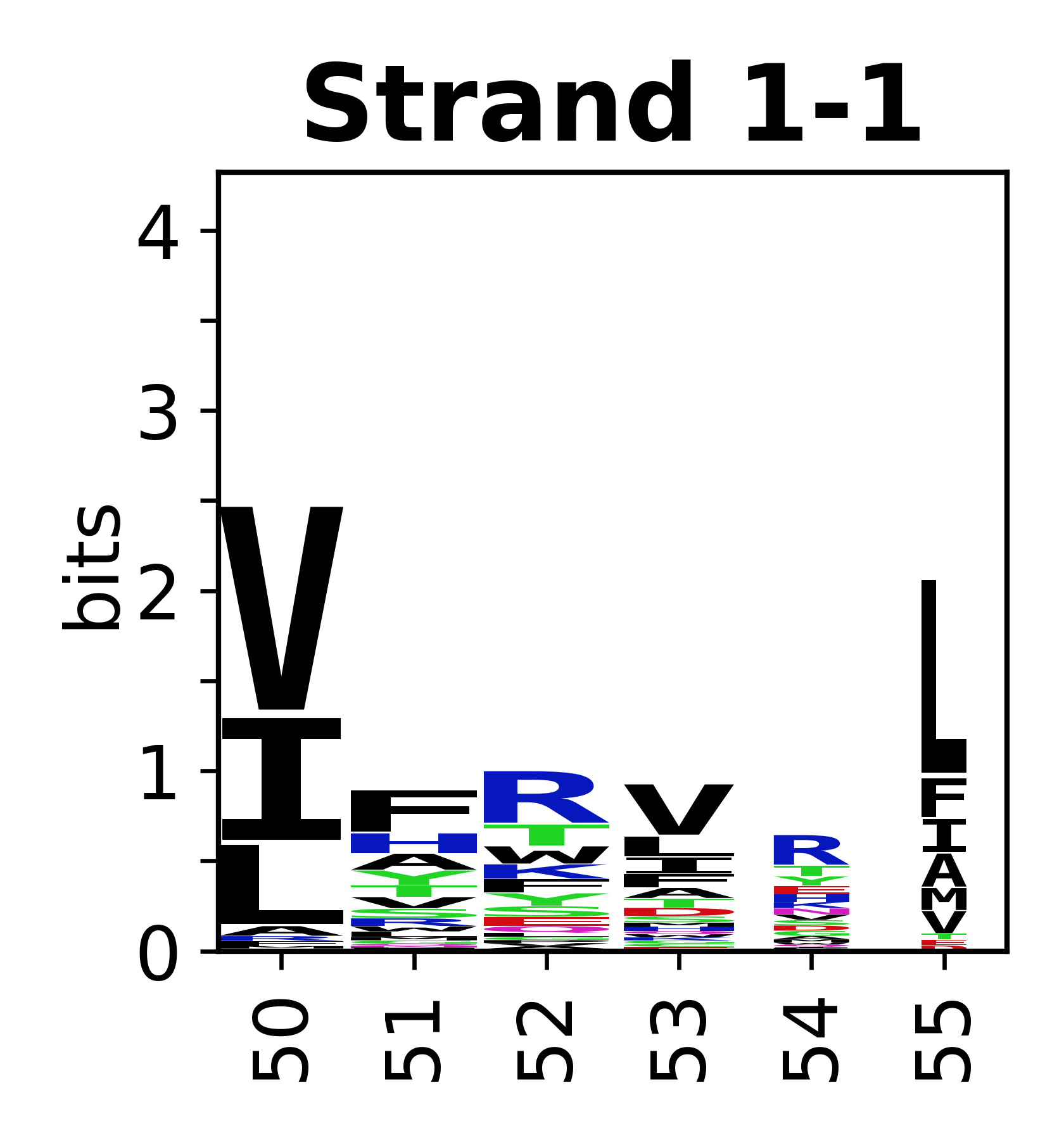
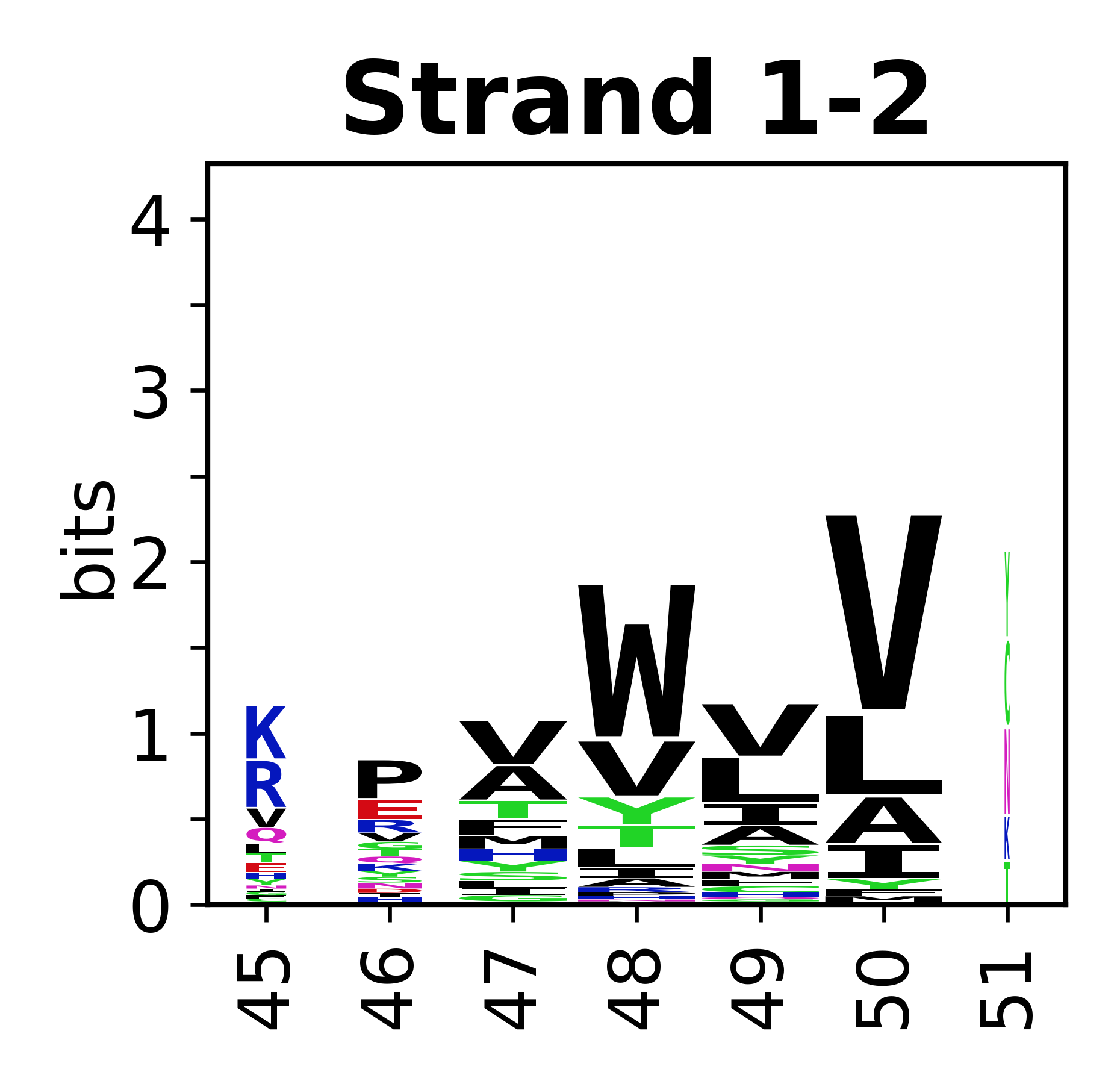
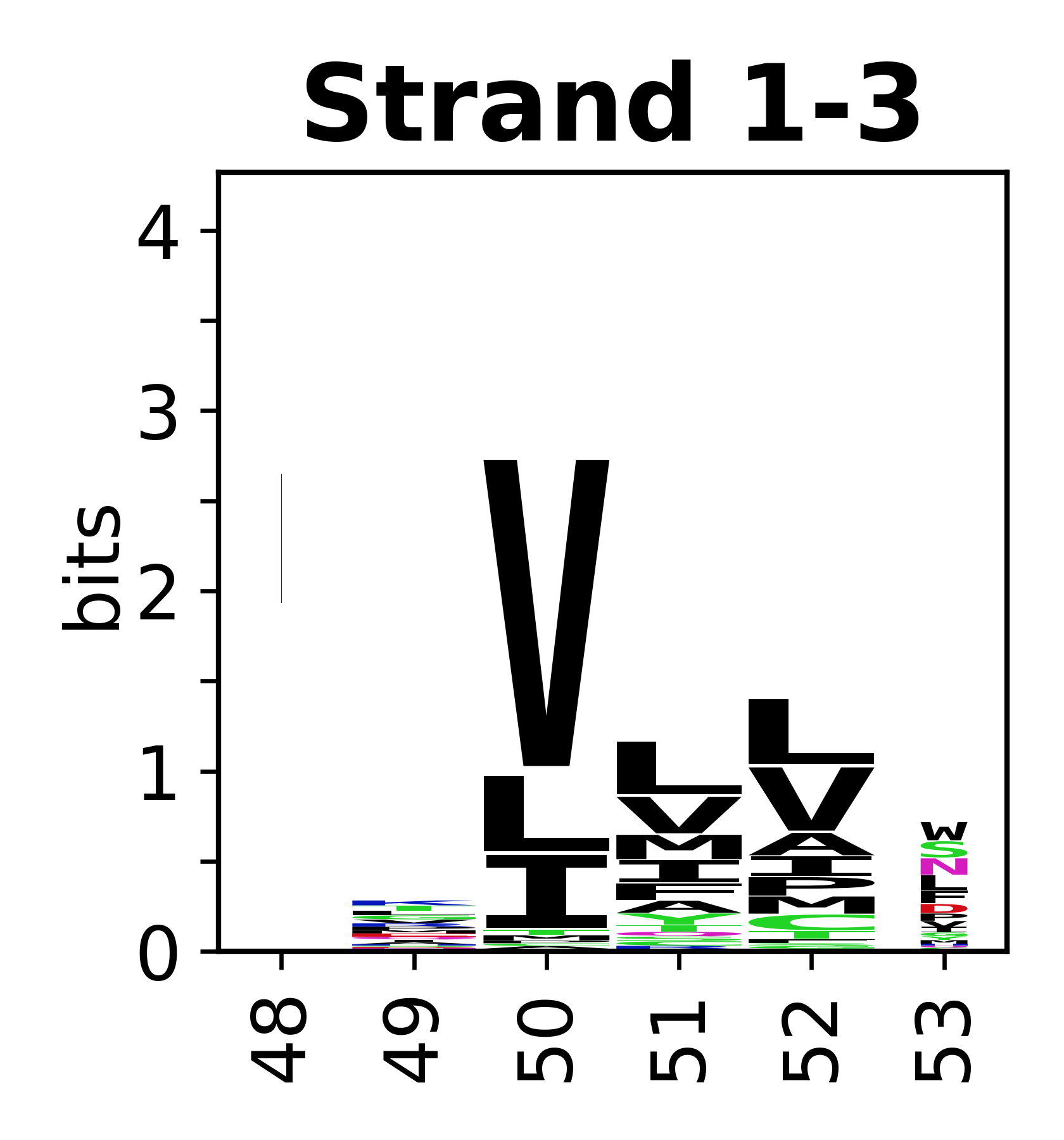
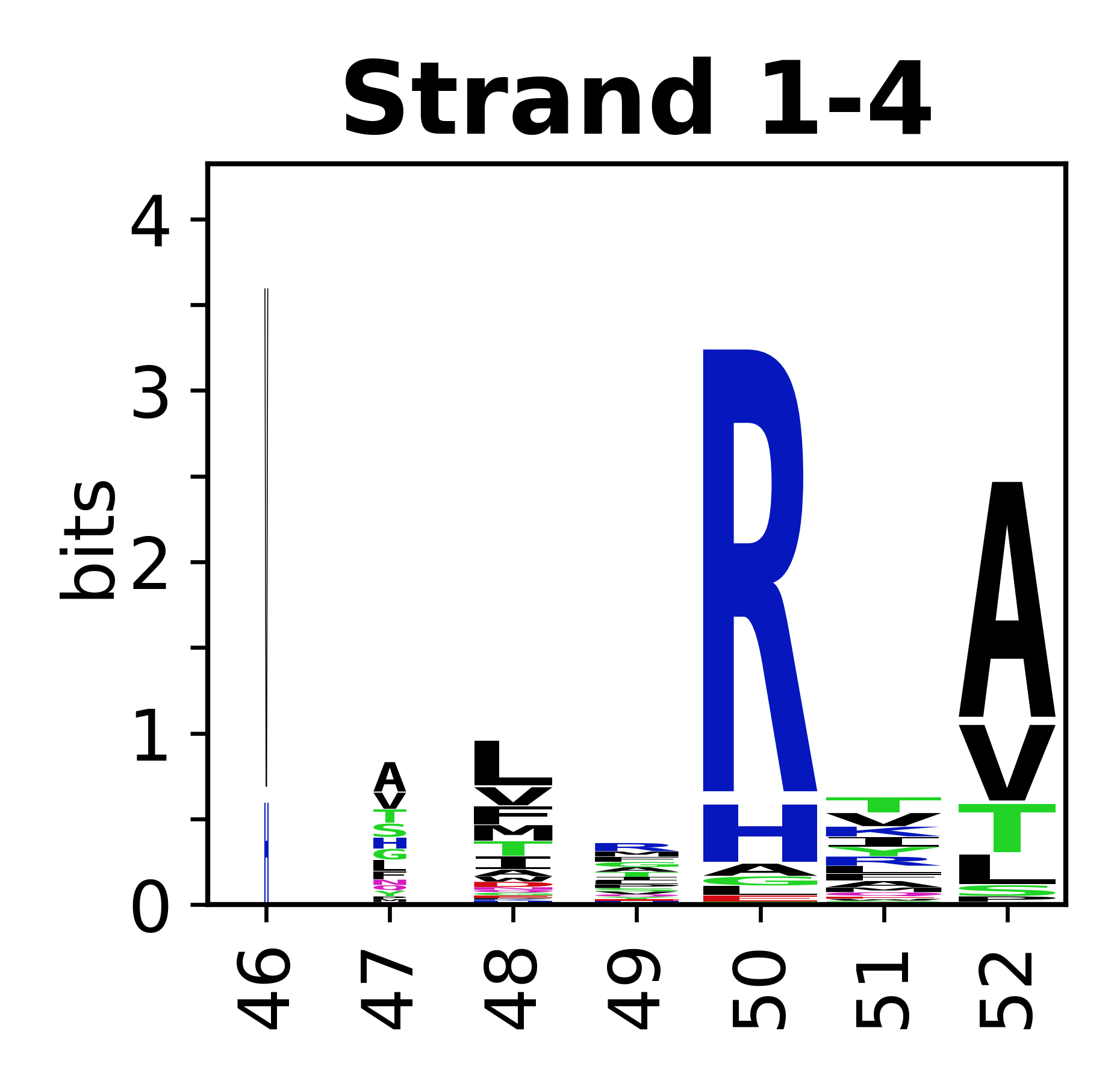
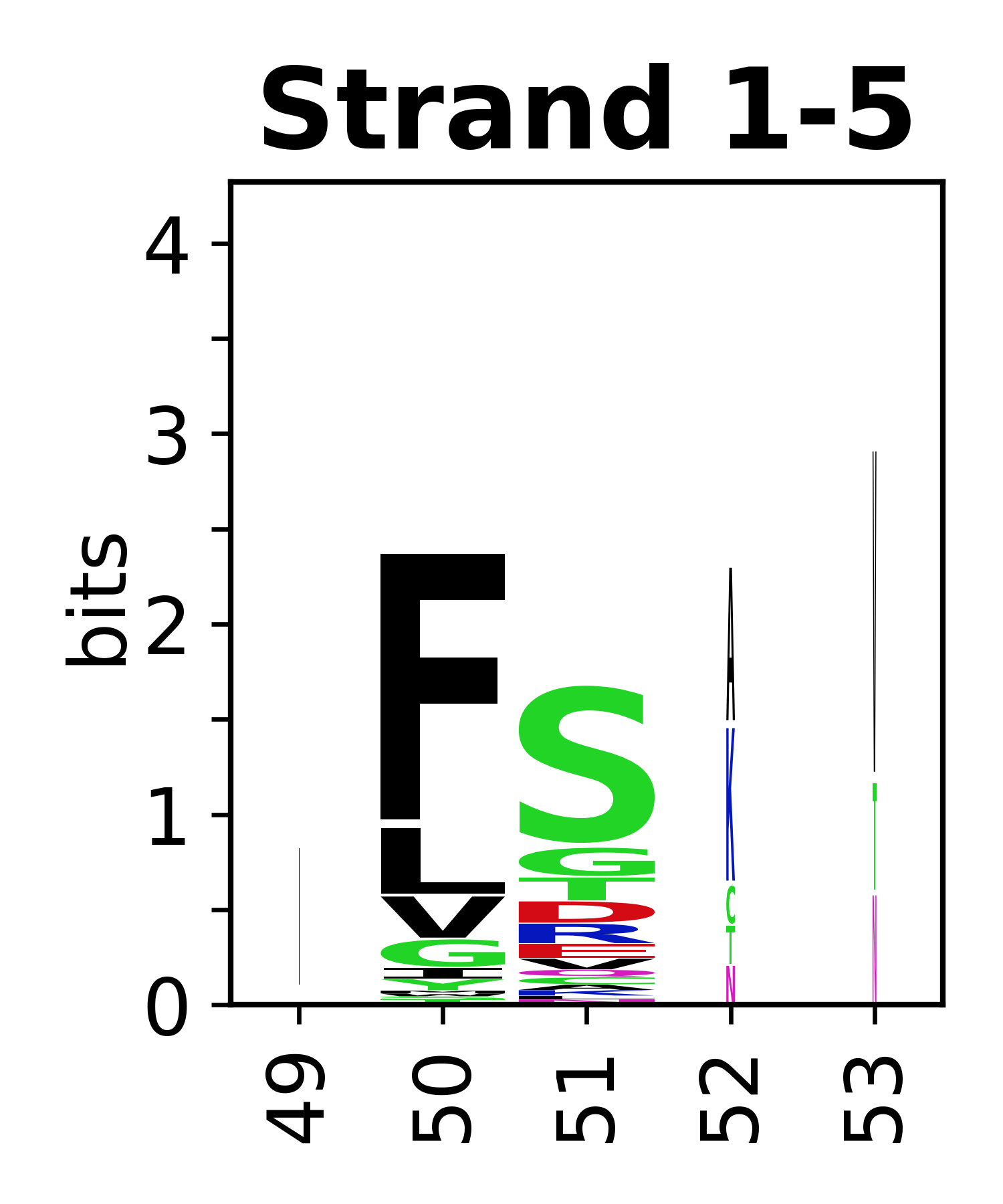
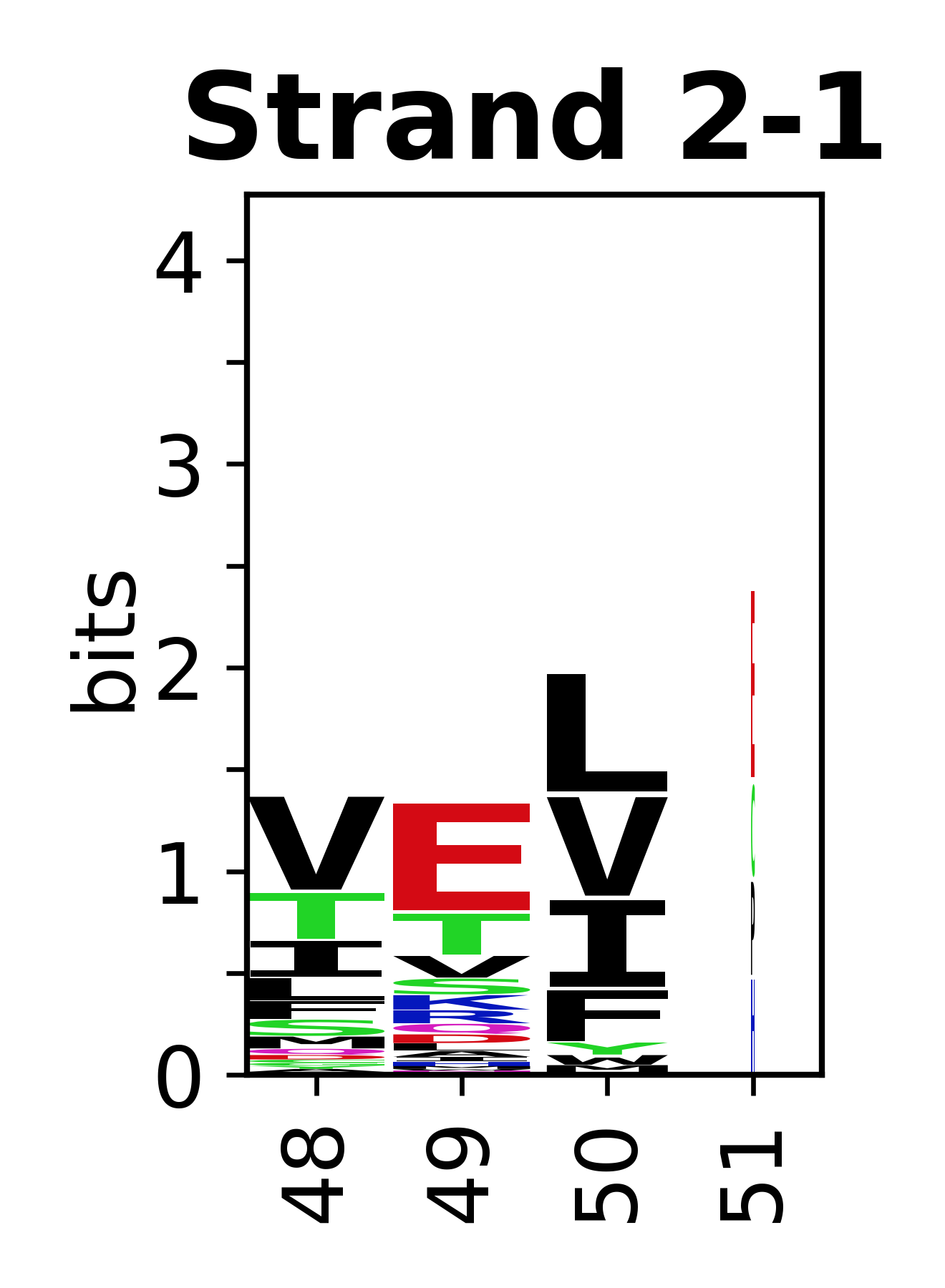
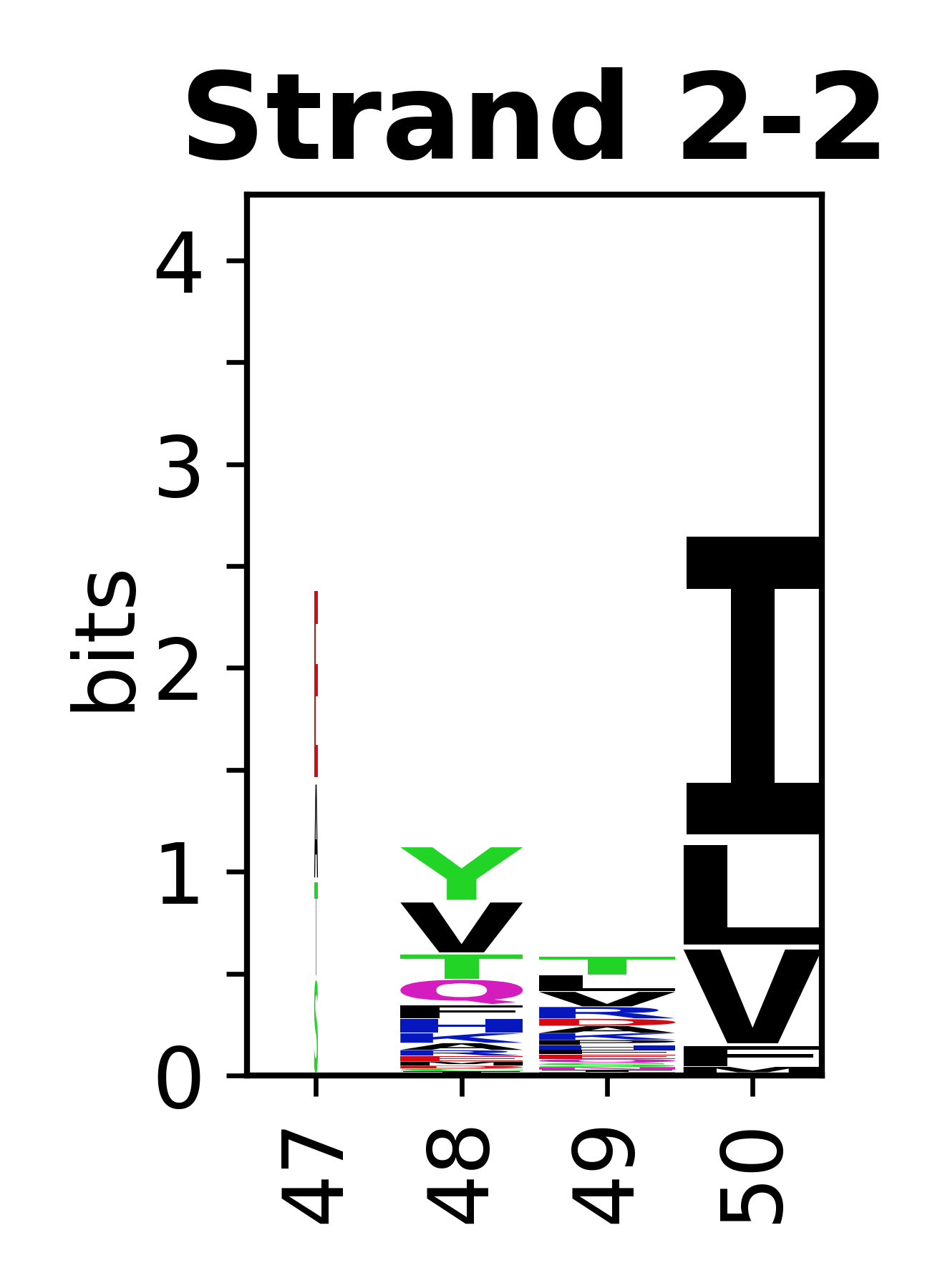
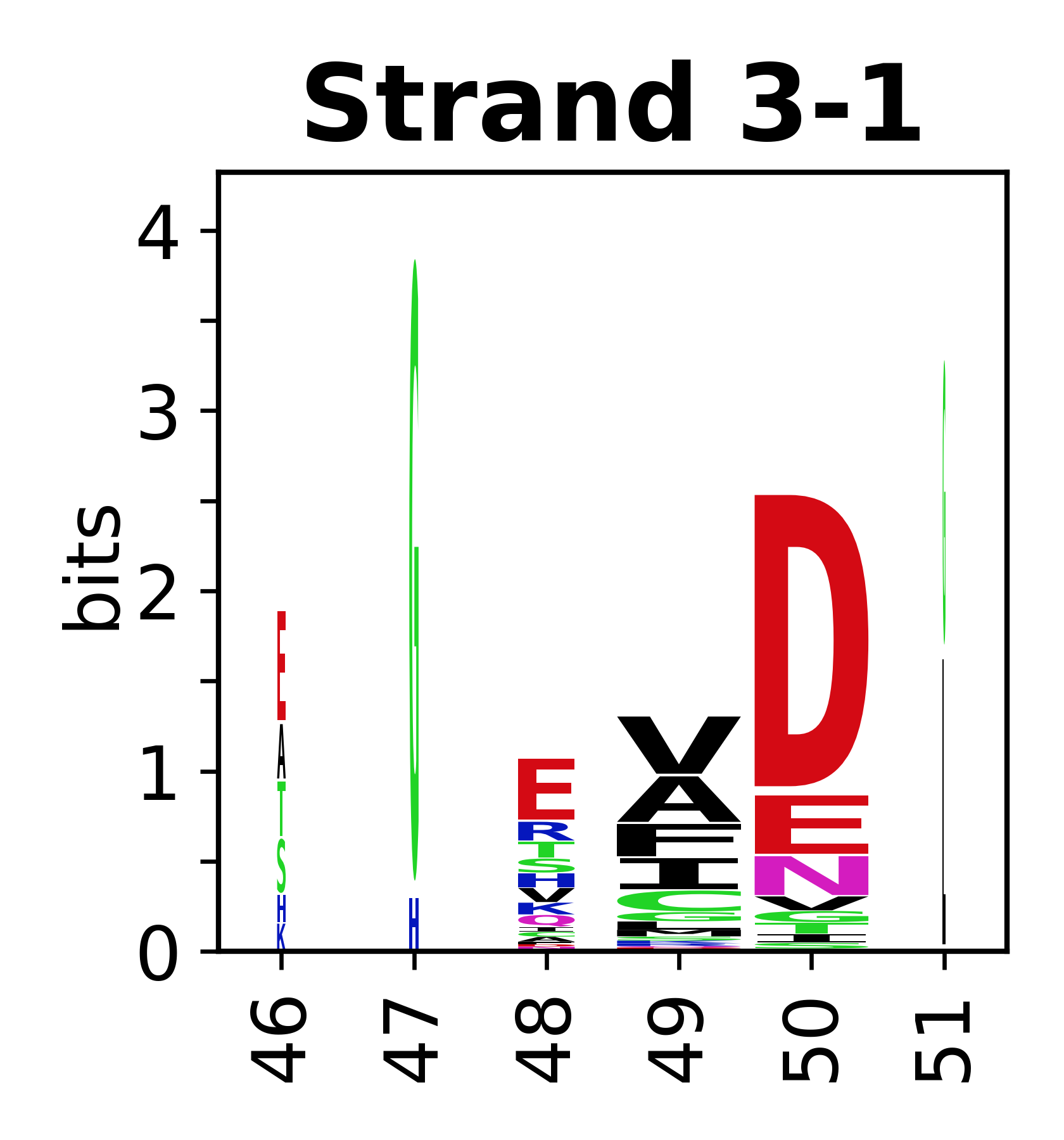
Sequence of SSEs
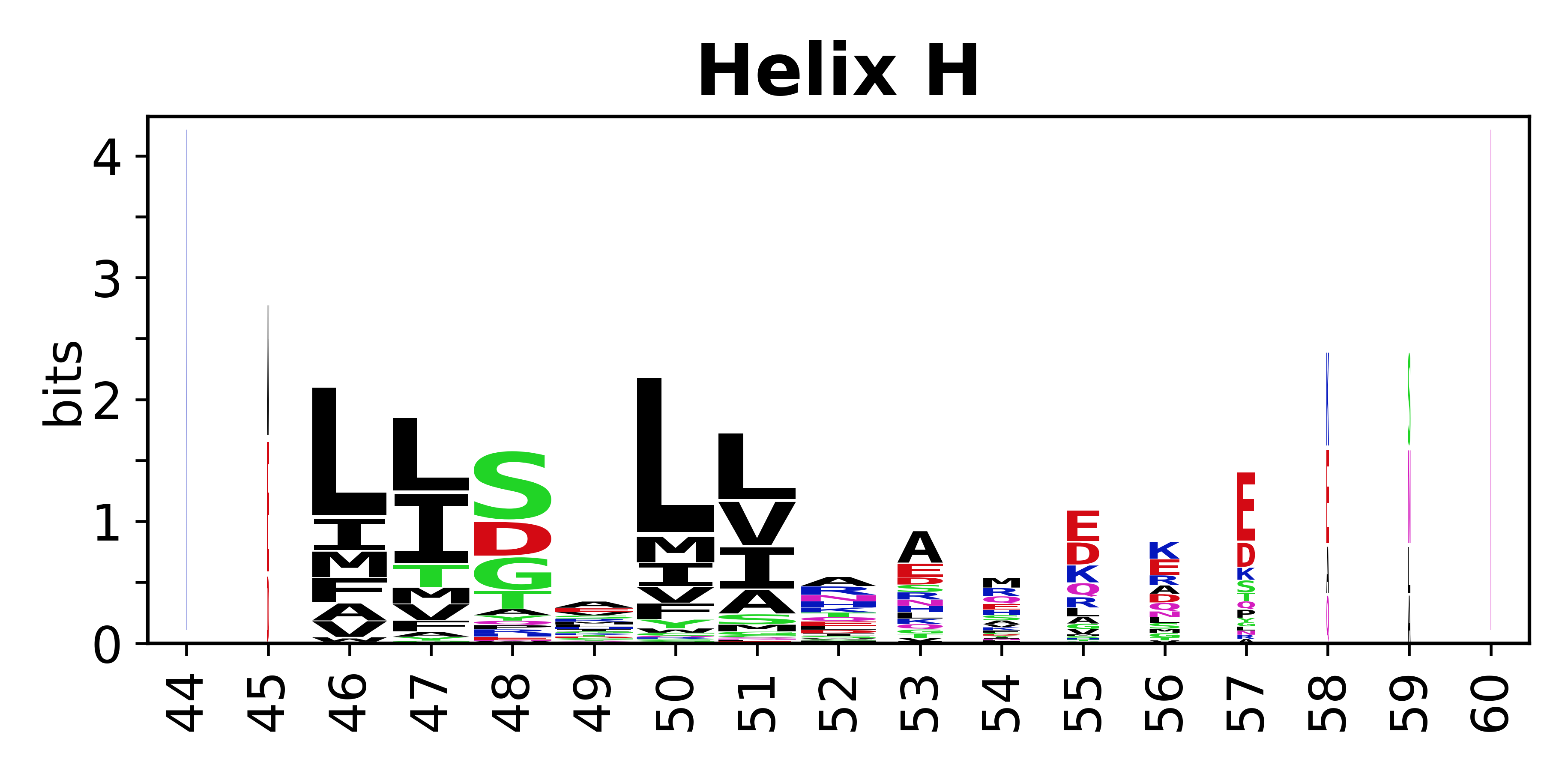
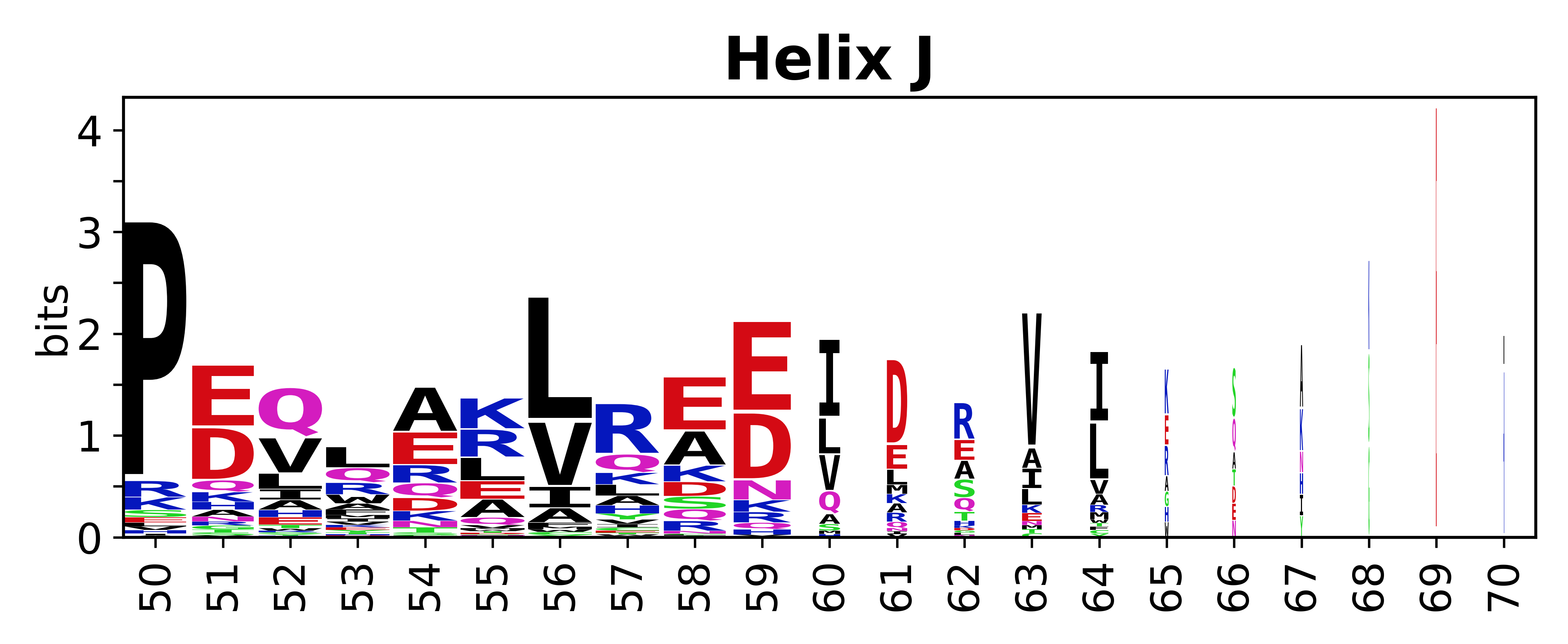
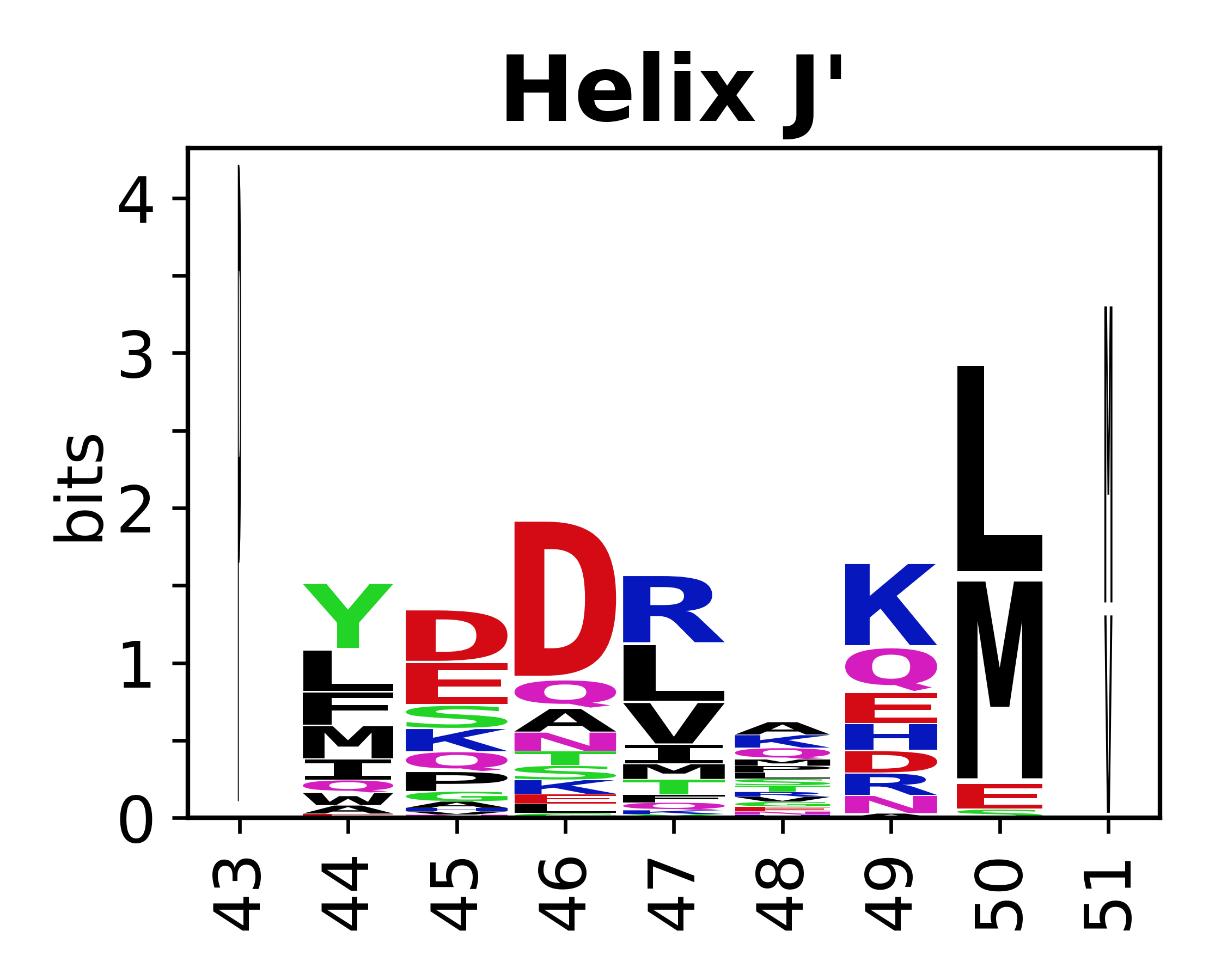
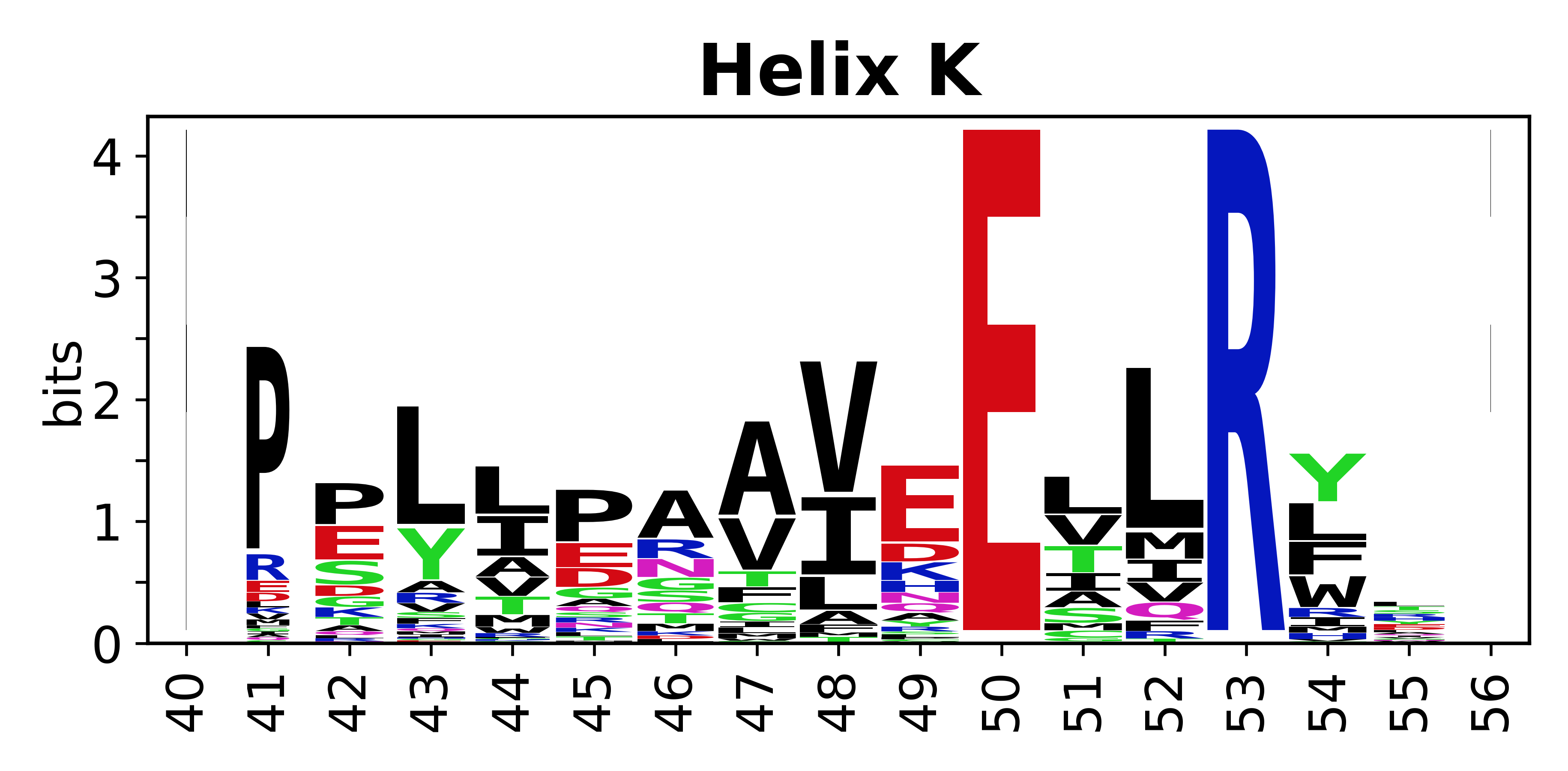
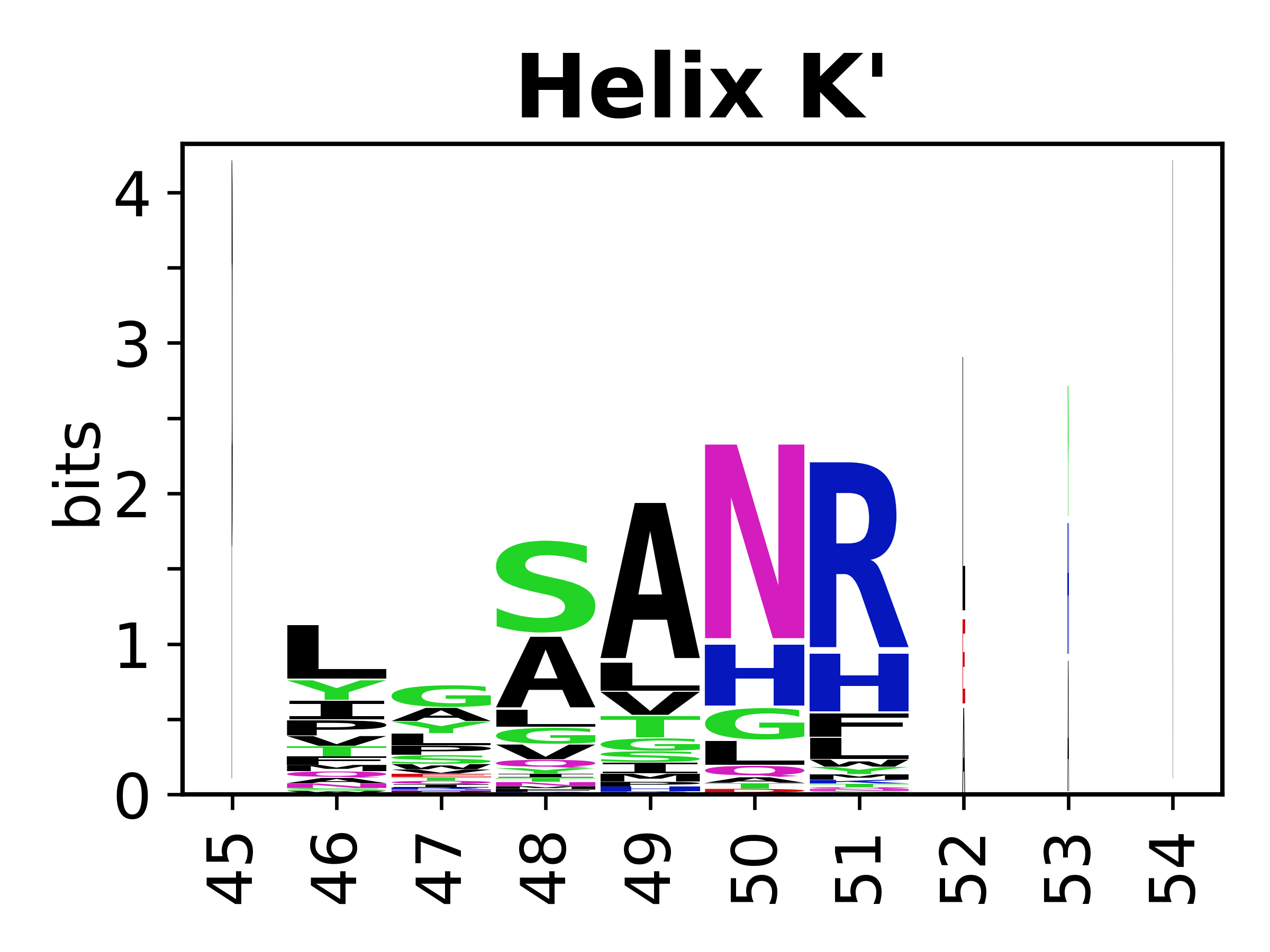
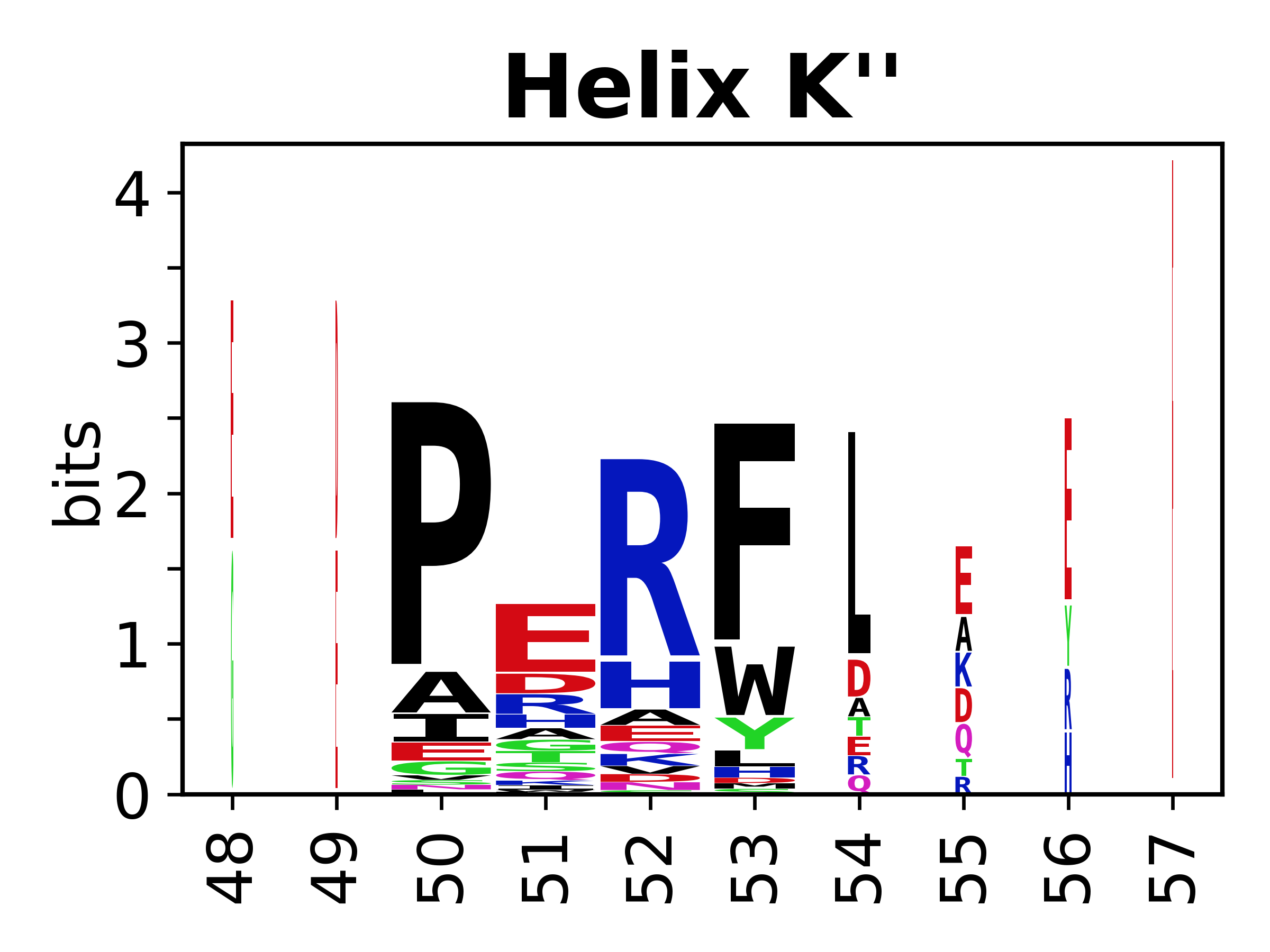
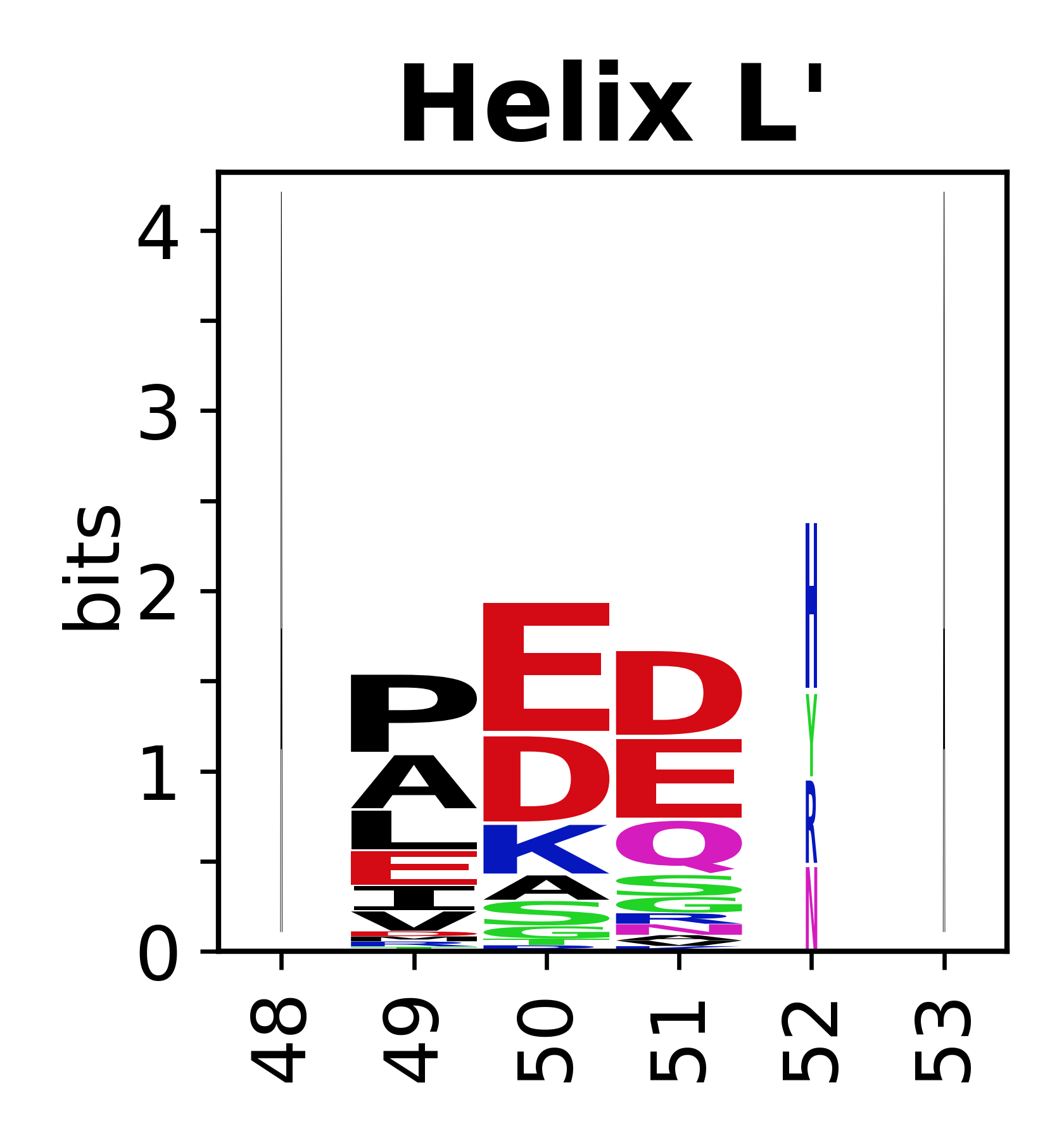
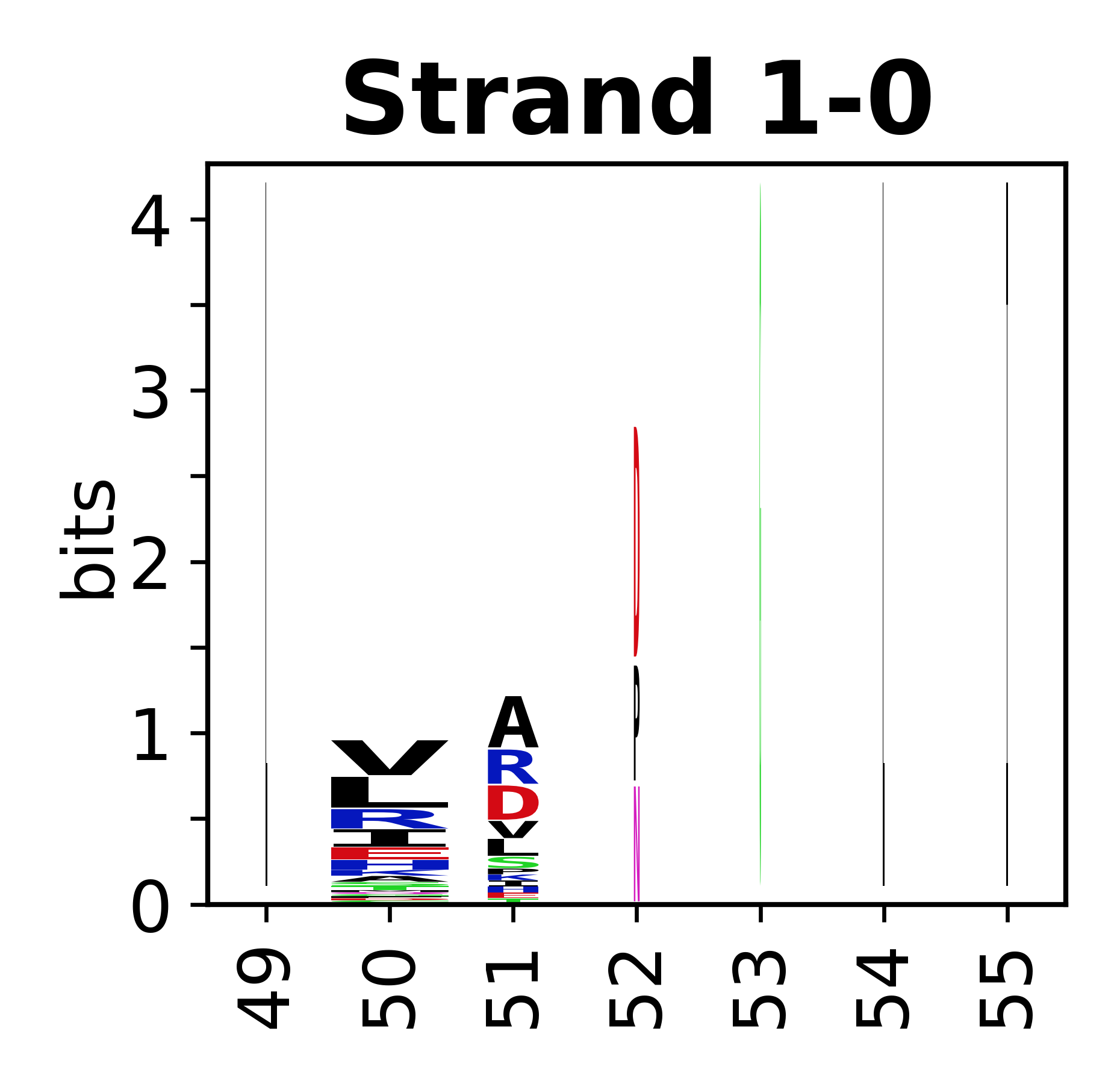
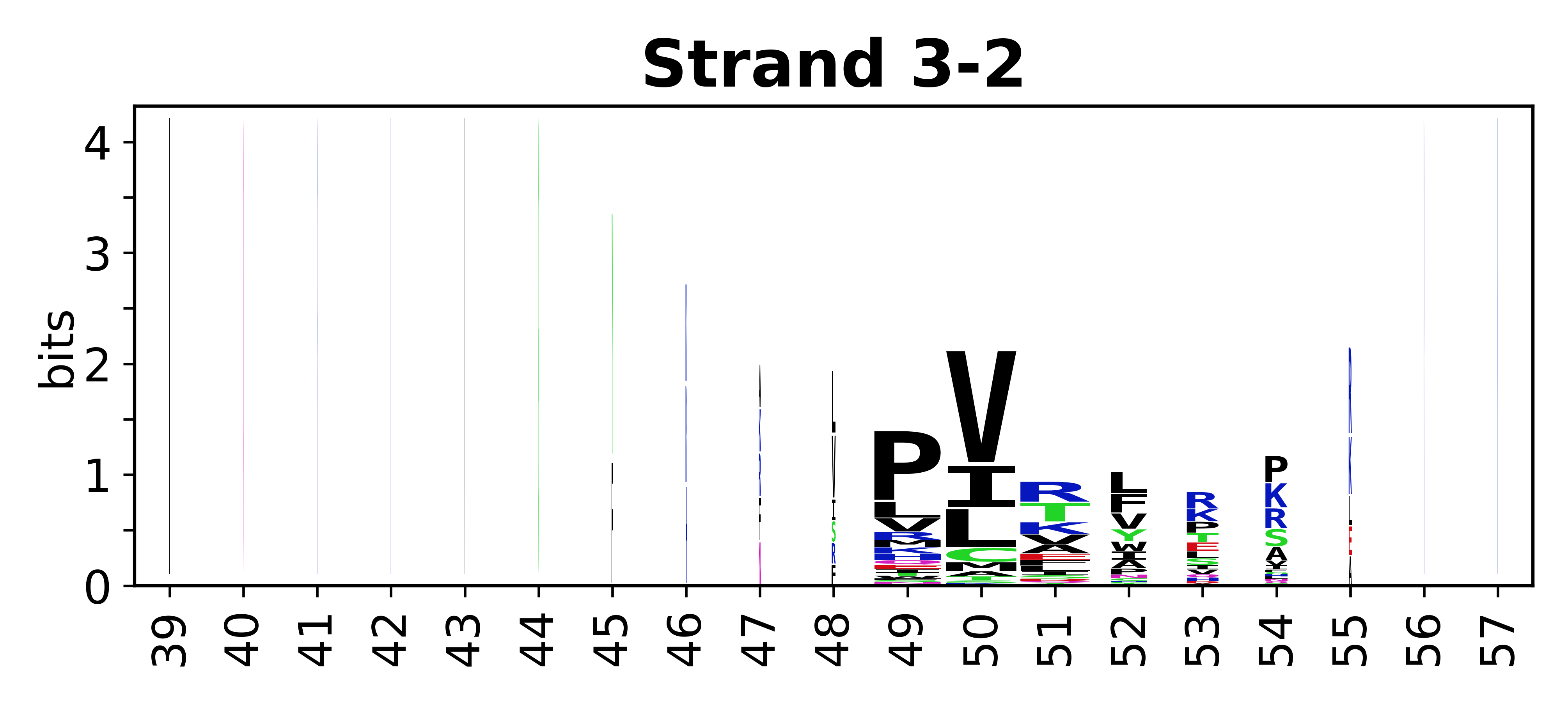
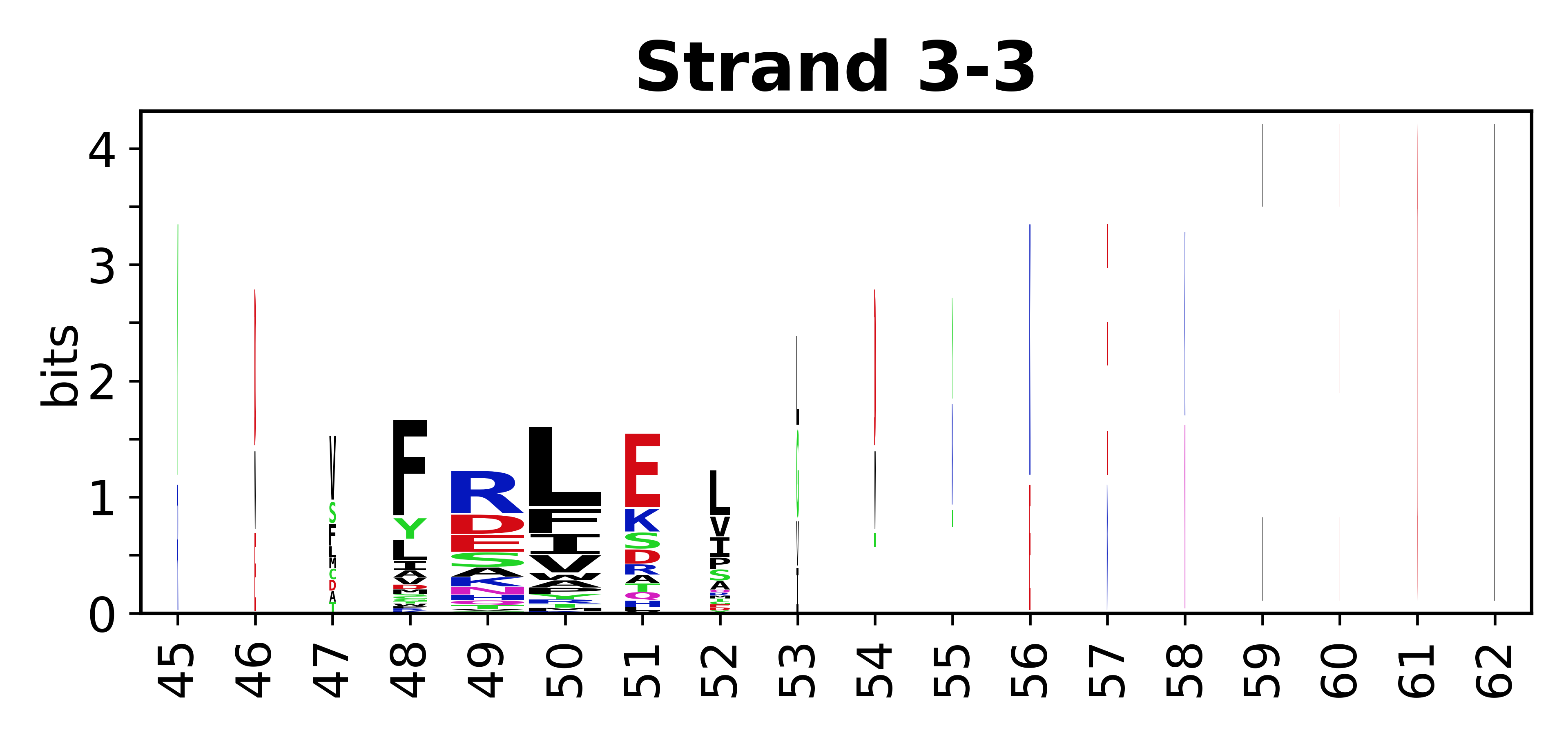
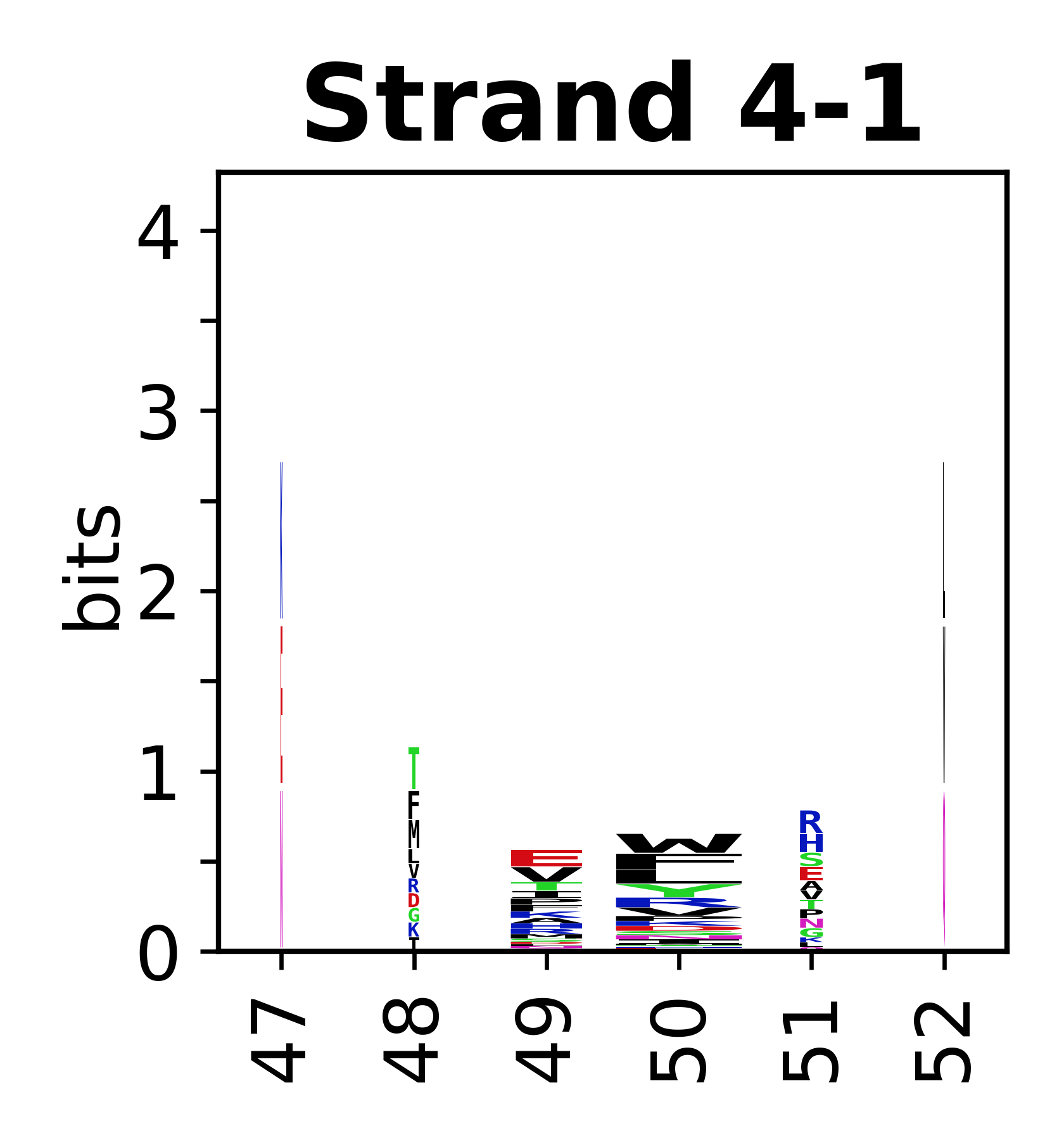
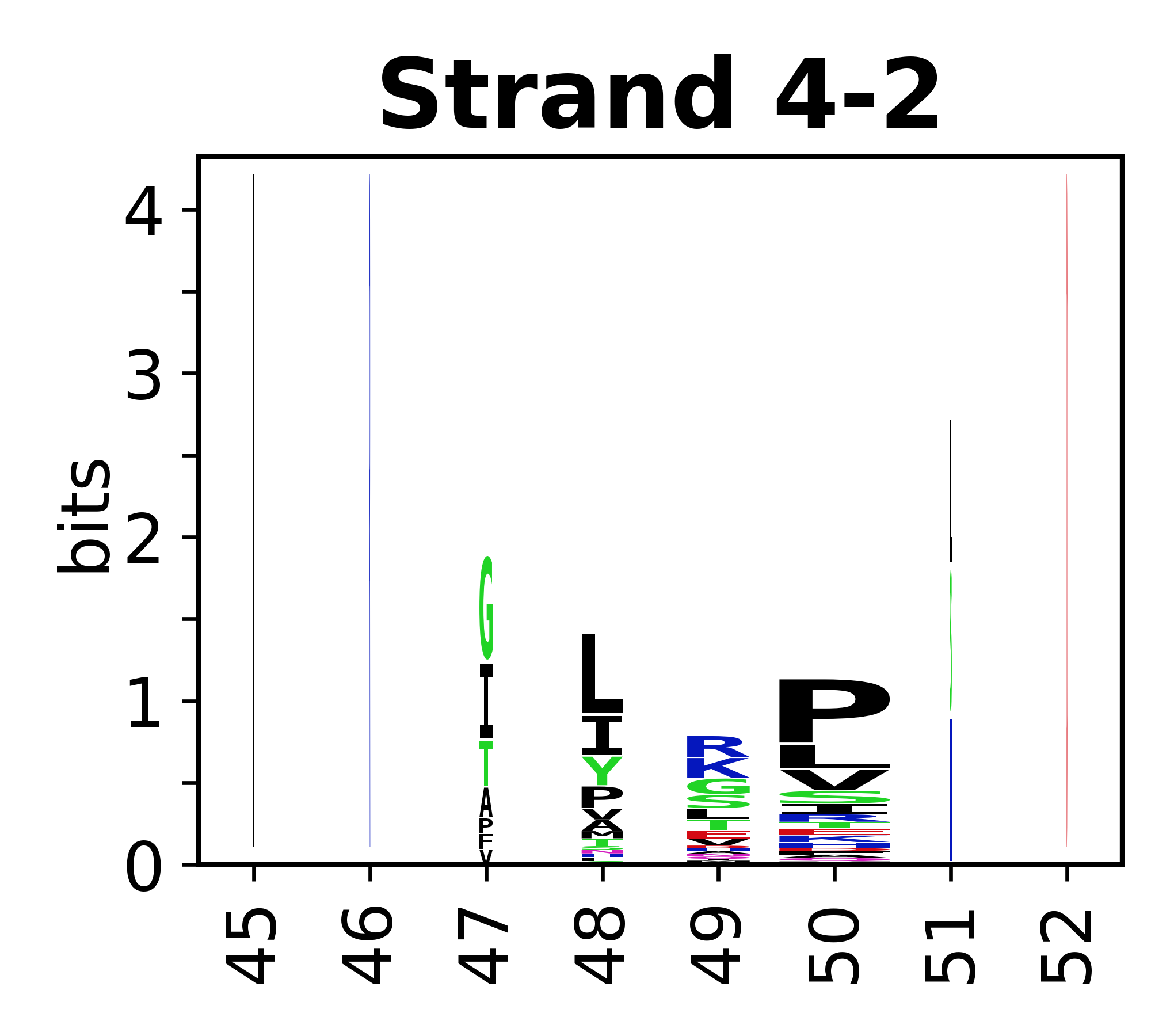
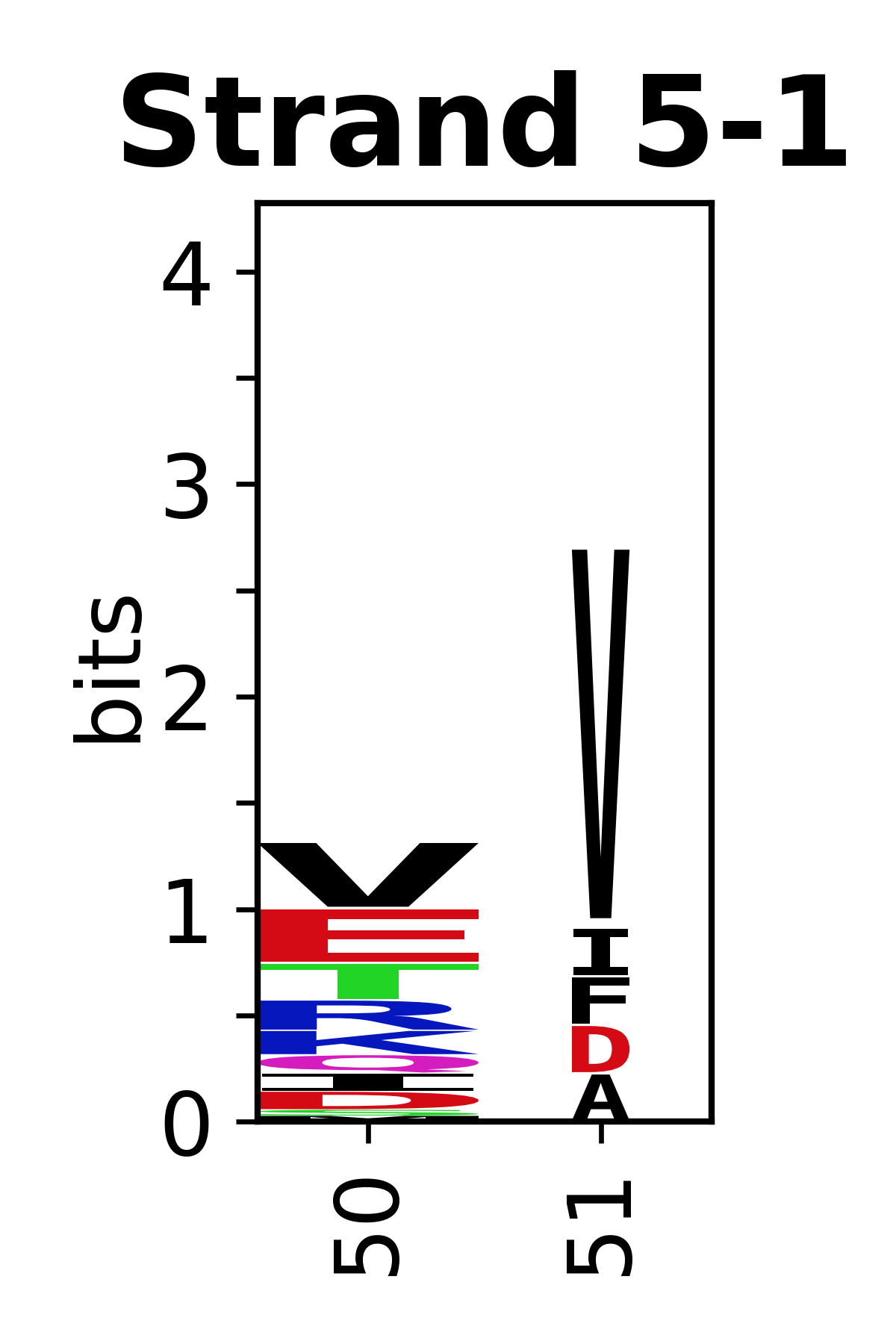
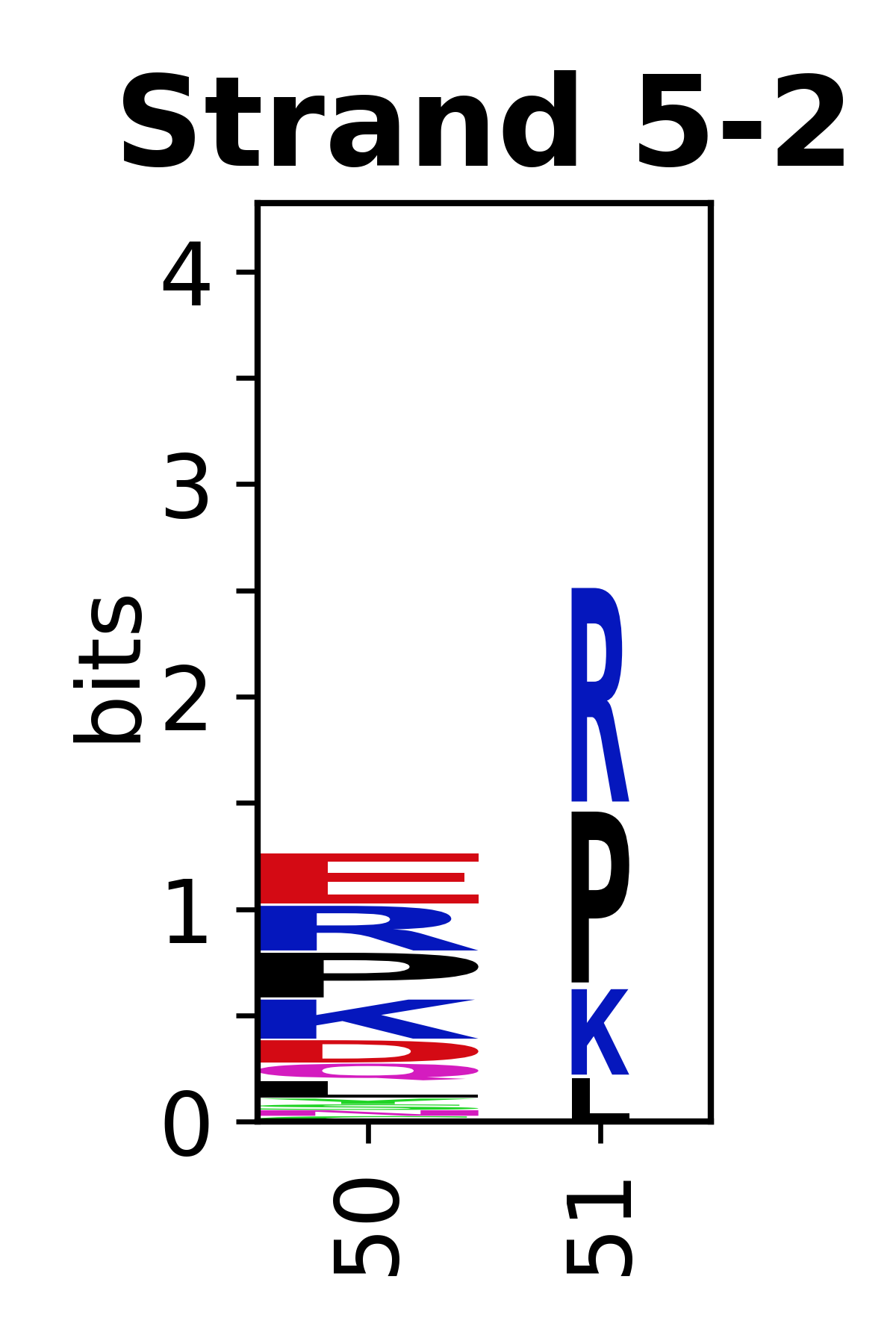
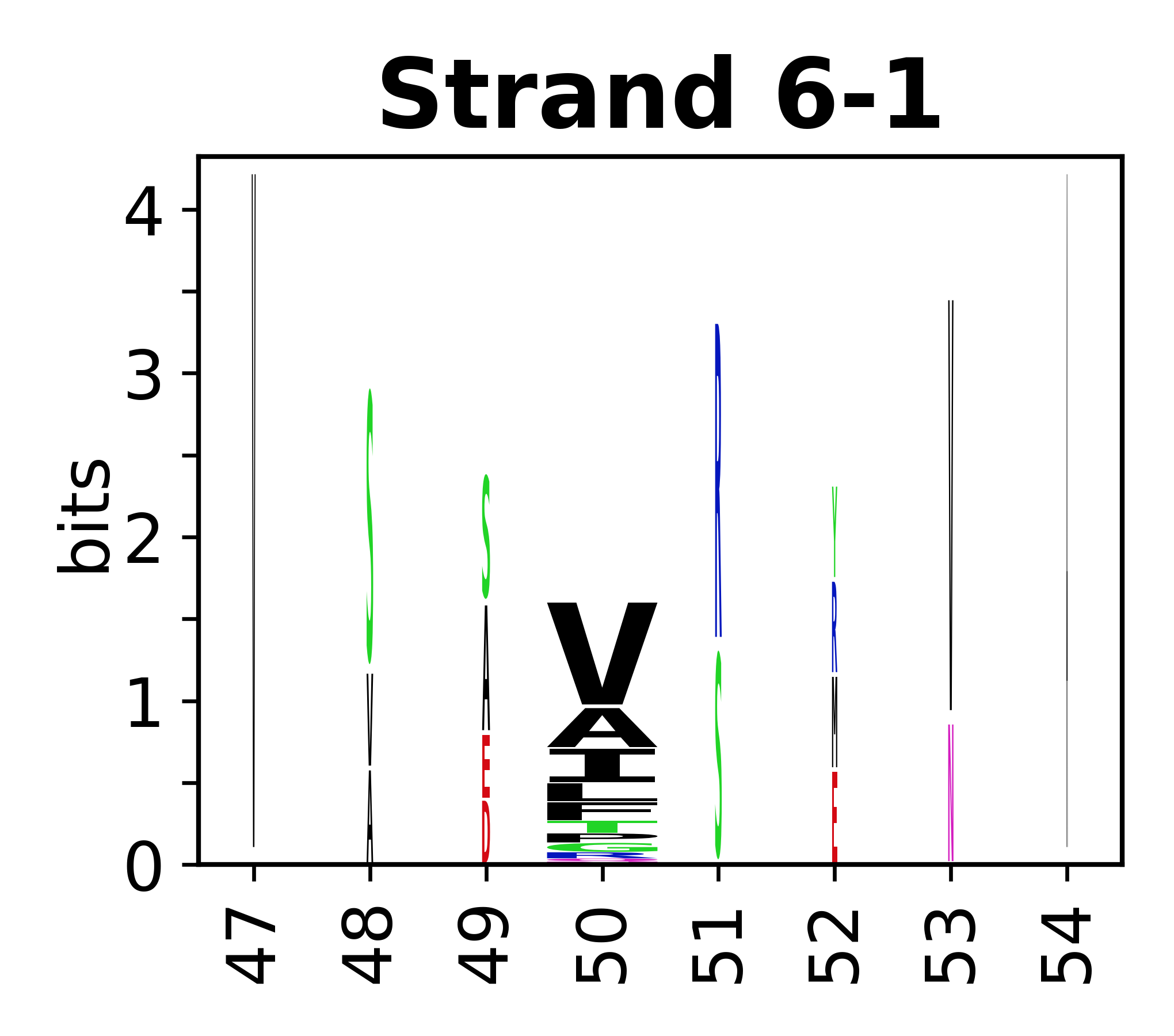
The amino acid sequences for each SSE can be aligned and used to produce a sequence logo. Where the sequence conservation is sufficient, we can establish a generic numbering scheme: the most conserved residue in helix X serves as its reference residue and is numbered as @X.50. The remaining residues in the helix are numbered accordingly.
- Helices
-
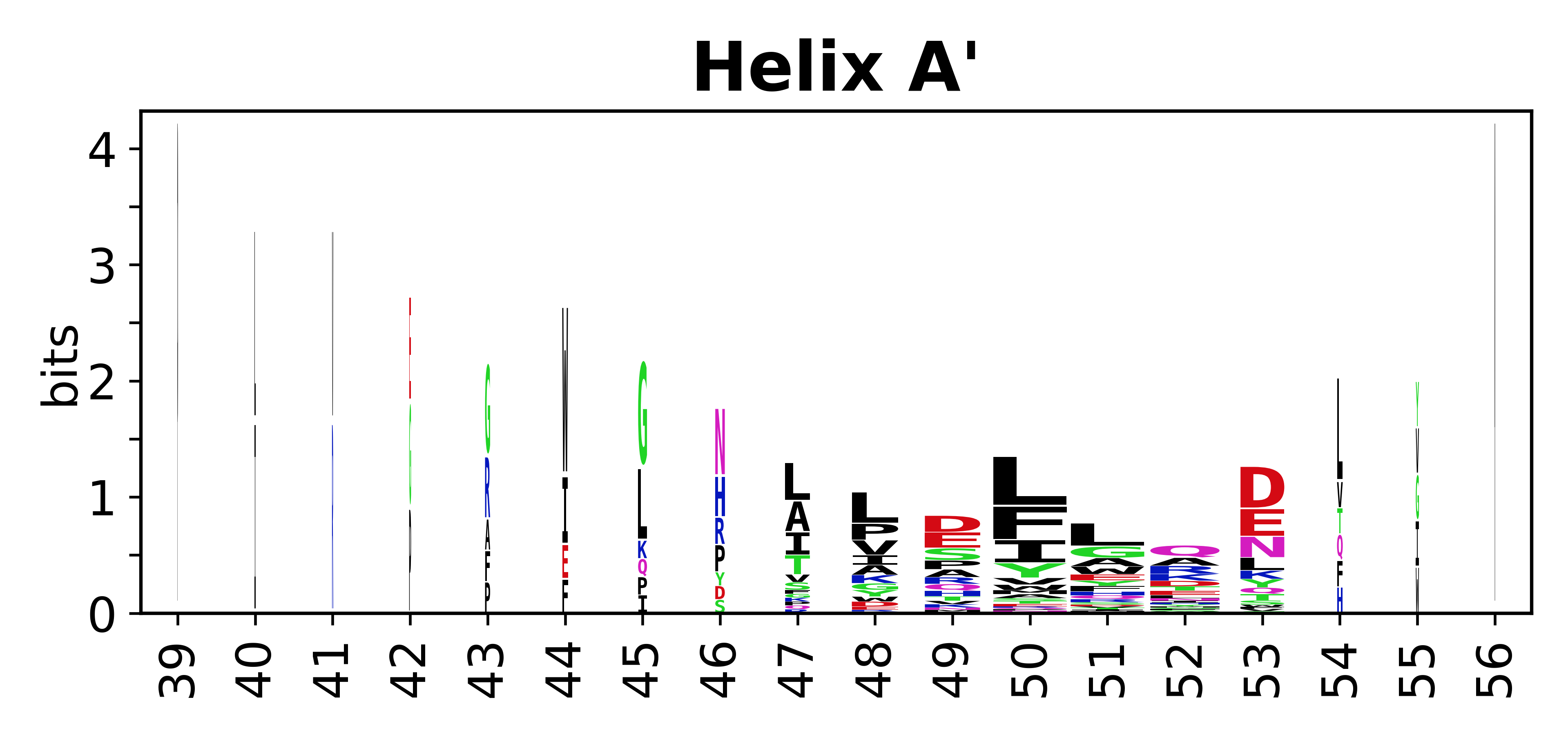
-

-
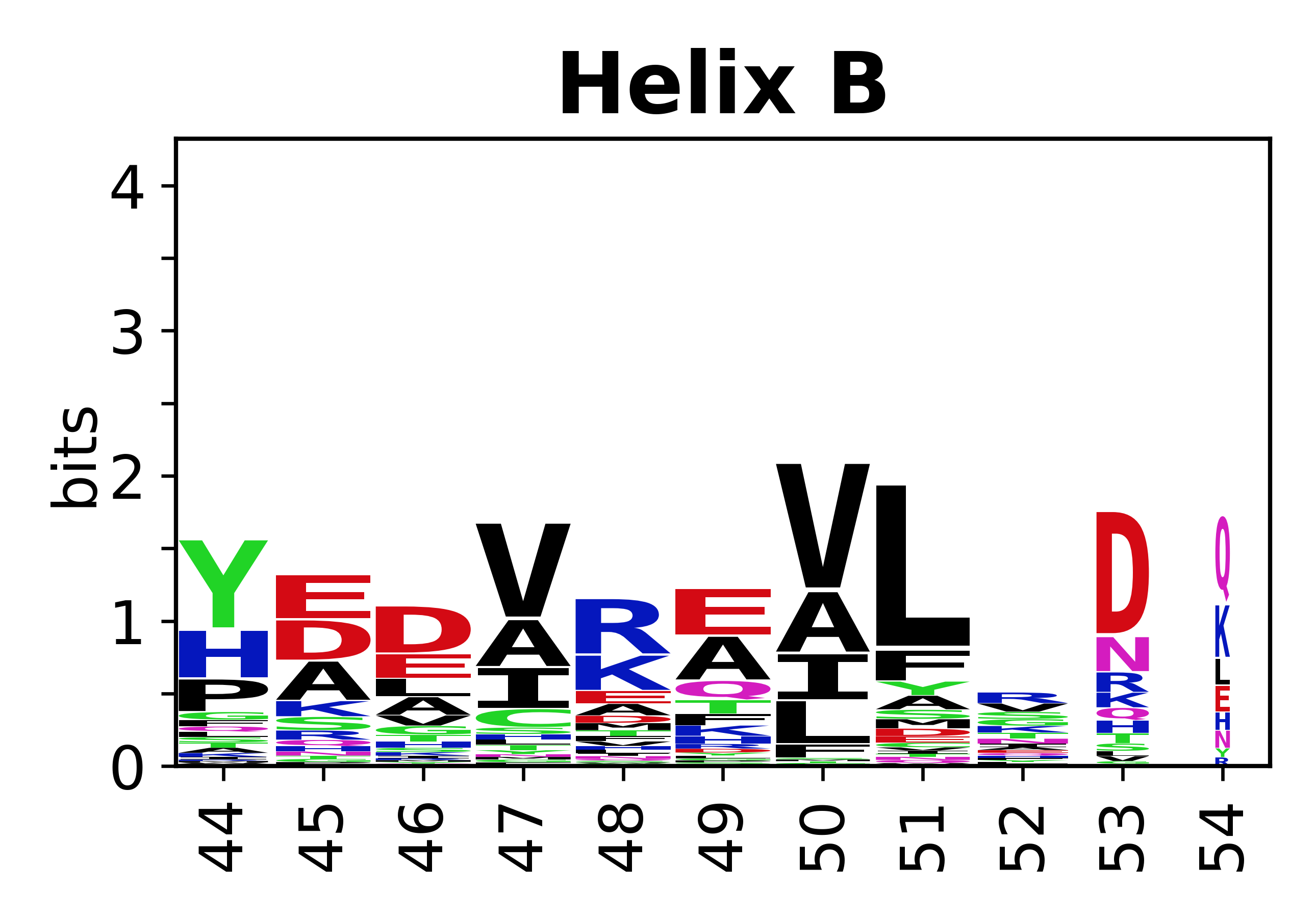
-
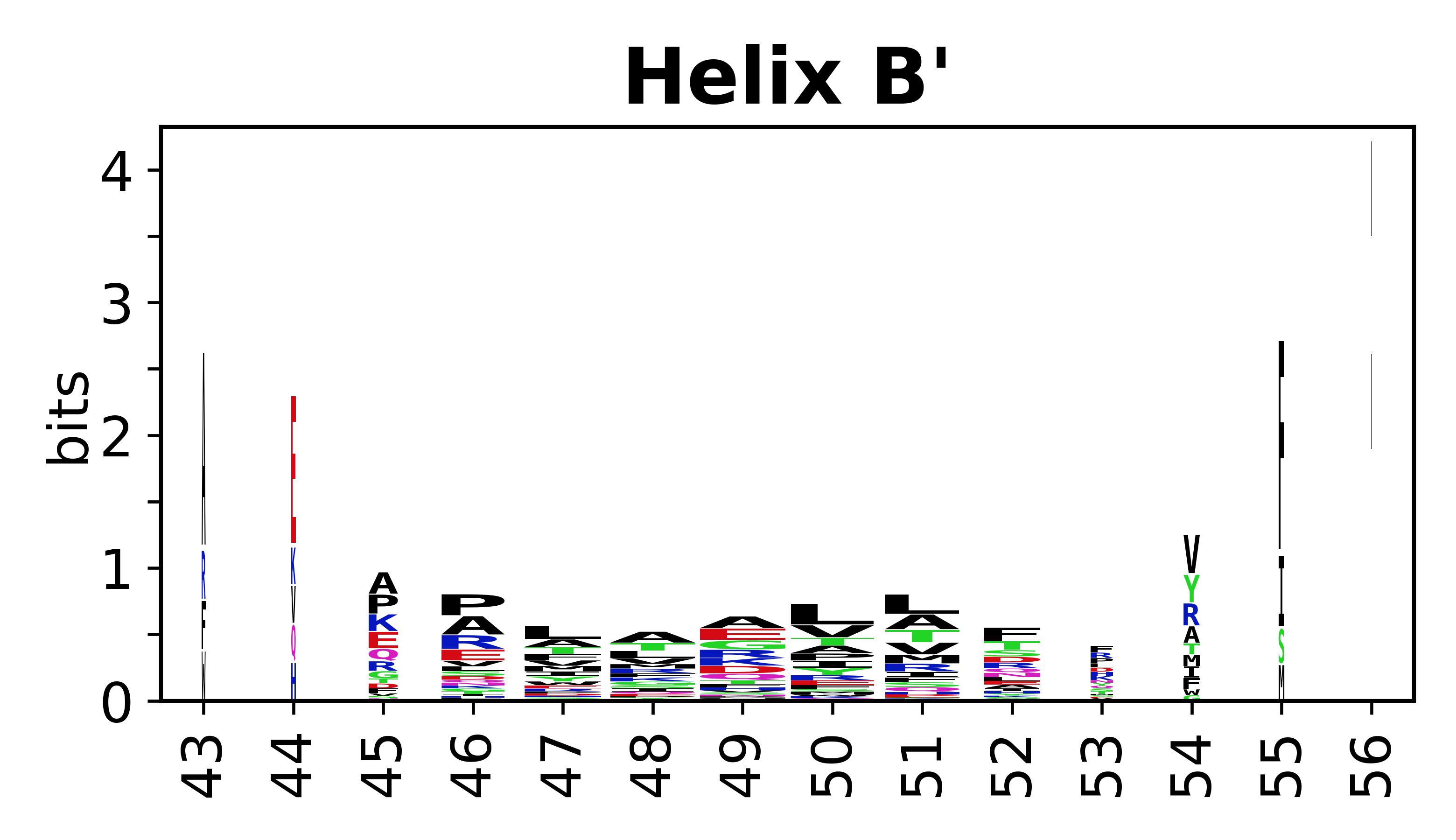
-
-
-
-
-
-
-
-
-
-

-
-
-
-
-
-

-
- Beta strands
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-